How to tell if child has mitochondrial disorder
Mitochondrial Disease | Boston Children's Hospital
How are mitochondrial disorders diagnosed?
Because symptoms are so varied, affecting multiple organs in the body, diagnosing mitochondrial disease can be challenging. Sometimes other disorders not involving the mitochondria are mistakenly diagnosed as mitochondrial disease. The opposite is true as well: Sometimes individuals who truly have mitochondrial disease are diagnosed as having something else.
Many symptoms of mitochondrial disease, such as failure to thrive, short stature, poor stamina, developmental delay, seizures, poor muscle tone, vomiting, severe constipation, or diarrhea, have a variety of other causes. Because of this, it’s usually not a single symptom but a combination of two, three or more different symptoms that leads clinicians to suspect mitochondrial disease.
Genetic testing is the most reliable way to diagnose and categorize a mitochondrial disorder. We may recommend genetic testing for your child (and sometimes for parents too) if any of these symptoms are present:
- developmental delay with involvement of other organs
- dardiomyopathy (disease of the heart muscle) or unexplained heart block, or impaired electrical signals in the heart
- high levels of lactate in the blood or cerebrospinal fluid, when other symptoms are present
- certain abnormal findings on brain imaging
- ophthalmoplegia (impaired eye movement) or ptosis (drooping upper eyelid)
- hearing loss
- severe gastrointestinal dysmotility (weak or lost muscular contractions in the intestines) or intestinal pseudo-obstruction (bowel obstruction caused by inability of the intestine to push food through)
- significant developmental regression in the setting of an illness
Genetic testing often begins with analyzing the mitochondrial DNA and, if results are negative, testing the nuclear DNA for genes known to be involved in mitochondrial disease. If these tests come up negative, the child’s nuclear DNA may need to be completely analyzed through whole exome sequencing (this is similar to whole genome sequencing, but analyzes just the genes that code for proteins).
The type and depth of genetic testing we recommend will depend on the child’s symptoms and how strongly we suspect mitochondrial disease. Unfortunately, at this writing, extensive genetic testing is not always covered by insurance. If you or your child is in need of testing, we will work with your insurance companies to help with the process of approval.
Additional, non-genetic tests also help in diagnosing mitochondrial disorders. They include:
- biochemical tests on urine, blood and spinal fluid
- a muscle biopsy to examine the mitochondria and test enzyme levels
- magnetic resonance imaging (MRI) of the brain and spine
If a mitochondrial disorder is suspected or identified, we may refer your child for further testing and consultations with specialists.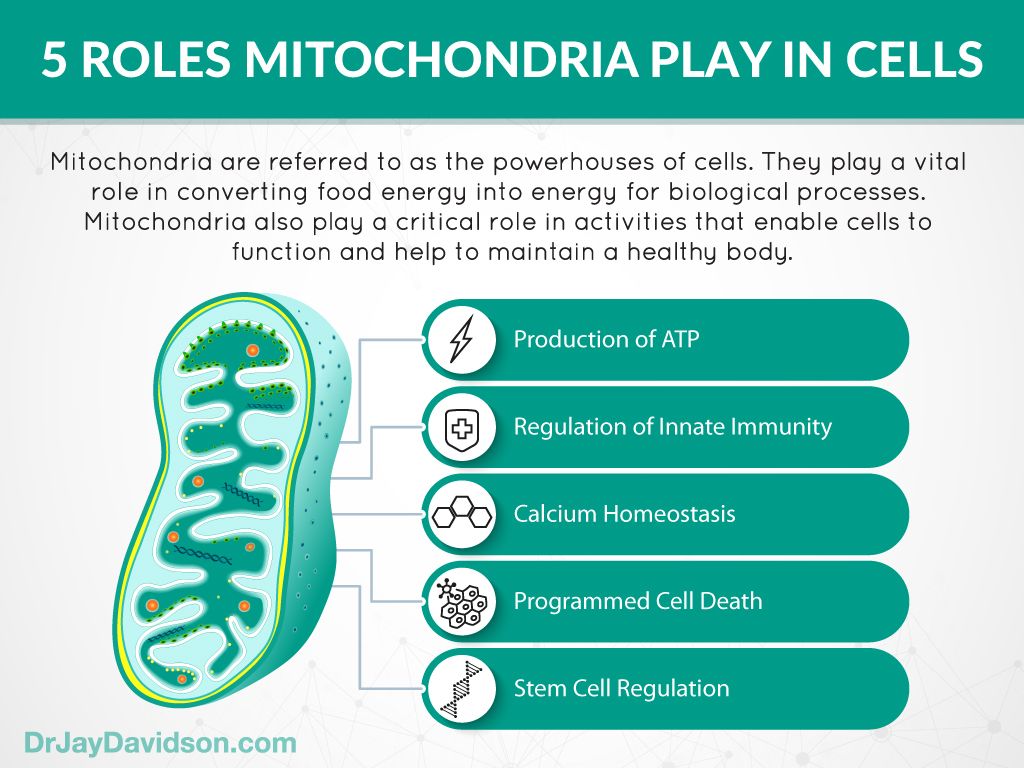 Testing depends on symptoms, and may include:
Testing depends on symptoms, and may include:
- echocardiogram
- electrocardiogram (EKG)
- eye examinations
- hearing tests
Managing your child's condition
While mitochondrial disorders have no cure, children often do better when symptoms are caught and addressed early. Your child will be followed closely and screened for a variety of conditions related to mitochondrial disease, such as heart, vision, and hearing problems. Symptoms can often be alleviated by maintaining good general health, including careful attention to nutrition and avoiding infections and dehydration.
Exercise is one of the few proven methods for improving mitochondrial functioning and can help preserve your child’s strength and endurance. Exercise regimens should be supervised, should build up very gradually in intensity and should be suspended during illness.
What are the treatment options for mitochondrial disorders?
Currently, vitamins and supplements are the mainstay of treatment for mitochondrial disorders. They are thought to help mitochondria produce energy and reduce accumulation of toxic compounds inside cells. These supplements and cofactors are sometimes given together in “cocktails.” At present, the compounds that have been most well tested include coenzyme Q10 and creatine, but we also use a great variety of other supplements.
They are thought to help mitochondria produce energy and reduce accumulation of toxic compounds inside cells. These supplements and cofactors are sometimes given together in “cocktails.” At present, the compounds that have been most well tested include coenzyme Q10 and creatine, but we also use a great variety of other supplements.
Regimens for specific mitochondrial disorders may include compounds that are deficient because of the disease, such as arginine (in MELAS) and folinic acid (in Kearns-Sayre syndrome). At Boston Children’s, we are testing dichloroacetate (DCA), which counters the high levels of lactic acid found in some mitochondrial disorders. In certain disorders, we may prescribe a special diet.
Supportive care
Because children with mitochondrial disease tend to be very sensitive to the stresses caused by minor illnesses like colds and fevers, your clinician will sometimes prescribe additional supportive therapies when the child is sick. These can include vitamins, cofactors, medications to counter biochemical imbalances that may occur, beverages containing electrolytes and sometimes intravenous (IV) hydration and IV antibiotics.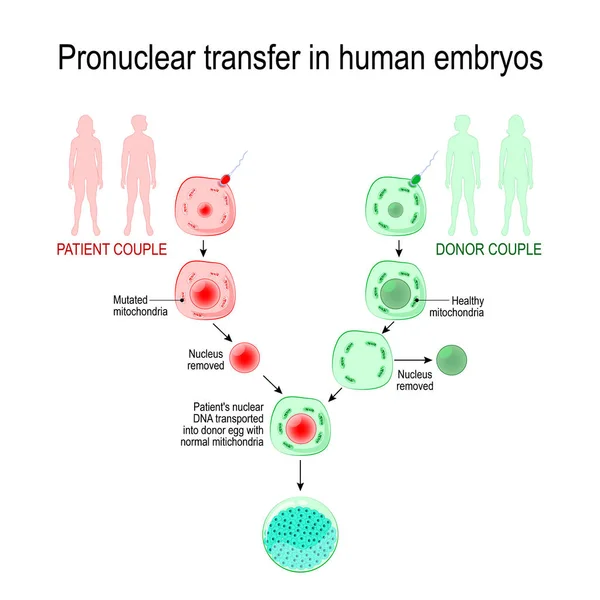 In some instances, your child may need to be monitored in the hospital.
In some instances, your child may need to be monitored in the hospital.
Children with mitochondrial disease also are known to be sensitive to a variety of drugs, as well as anesthetics used for surgical procedures.
Mitochondrial Disorders | Riley Children's Health
- Find a Doctor
- Request An Appointment
- On This Page:
- Treatments
- Key Points to Remember
- Support Services & Resources
- Locations
- Departments Treating This Condition
Mitochondria
are small,
energy-producing structures in cells. The mitochondria are sometimes called the
power plants of cells because they produce most of the energy your body needs
to function. A mitochondrial disorder occurs when there is damage to the
mitochondria, which depletes the cells’ energy.
The mitochondria are sometimes called the
power plants of cells because they produce most of the energy your body needs
to function. A mitochondrial disorder occurs when there is damage to the
mitochondria, which depletes the cells’ energy.
Mitochondrial disorders vary in severity and age of onset from one person to the next. They can present at any point in life, though they generally first cause symptoms in childhood. If your child has a mitochondrial disorder, the brain, muscles and gastrointestinal (GI) tract will most likely be affected because each of these body parts require high amounts of energy to function properly, though virtually any organ system can be affected. Mitochondrial disorders may be inherited in a number of ways, but the exact pattern in a given family may be difficult to determine.
Some symptoms of mitochondrial disorders include:
- Abdominal distention
- Blindness
- Cardiomyopathy
- Deafness
- Dementia
- Developmental delay
- Feeding problems
- Frequent infections
- Heart rhythm problems
- Increased fatigue
- Low muscle tone
- Migraine headaches
- Muscle weakness
- Nausea
- Seizures
- Severe constipation
- Sleep apnea
- Stroke-like symptoms
- Trouble moving the eyes
- Vomiting
Patients do not typically experience all of these symptoms. The most common symptoms are fatigue, low muscle tone, developmental delay and GI problems.
The most common symptoms are fatigue, low muscle tone, developmental delay and GI problems.
Diagnosis of Mitochondrial Disorders
The following exams and tests may be used to help determine if your child has a mitochondrial disorder:
- Physical exam
- Blood tests to determine elevated lactic acid or other biochemical abnormalities
- Specific mitochondrial and/or nuclear gene testing
- Magnetic resonance imaging (MRI) scan of the brain
- Electrocardiogram and echocardiogram
- Eye exam
- Neurological evaluation
- Hearing test
Treatments
Treatments
There is no cure for mitochondrial disorders. However, treatment may
help reduce symptoms and slow down the long-term decline in health. Your child's
doctor may recommend avoiding certain types of medicines that may increase
symptoms, such as sedation medication.![]() In addition, supplements such as carnitine and/or
coenzyme Q are often prescribed to help control the
symptoms caused by a mitochondrial disorder. Doctors hope to improve a patient's
quality of life by managing symptoms with these treatment methods.
In addition, supplements such as carnitine and/or
coenzyme Q are often prescribed to help control the
symptoms caused by a mitochondrial disorder. Doctors hope to improve a patient's
quality of life by managing symptoms with these treatment methods.
Key Points to Remember
Key Points to Remember
- A mitochondrial disorder occurs when there is damage to the mitochondria which depletes the cells’ energy and, therefore, depletes the body’s energy.
- Most mitochondrial disorders affect the brain, muscles and GI tract, but other organs can be affected.
- There is no cure for mitochondrial disorders. However, treatment with certain medicines may help improve symptoms.
Support Services & Resources
Support Services & Resources
Visit the following websites to learn more about how mitochondrial disorders are diagnosed and treated.
Riley at IU Health offers a broad range of supportive services to make life better for families who choose us for their children's care.
Learn More About Riley Support Services
Genetics Home Reference
This online resource from the U.S. National Library of Medicine provides more information about mitochondrial disorders.
United Mitochondrial Disease Foundation
The United Mitochondrial Disease Foundation offers support and information and promotes research for the cure and treatment of mitochondrial disorders.
Locations
Locations
Locations
In addition to our primary hospital location at the Academic Health Center in Indianapolis, IN, we have convenient locations to better serve our communities throughout the state.
Sort through 3 facilities offering Mitochondrial Disorders care by entering your city or zip below.
See all facilities →
Departments Treating This Condition
Departments Treating This Condition
90,000 fever and mitochondrial disease - Autism risk factors 04/18/13
Article of Dr. Robert Navier, a leading communication researcher between the immune system, anomalies of mitochondria and the risk of autism
Author: Robert Vesier / Robert K. Naviaux
9000 9000 9000 9000 9000 9000 9000 9000 Translation: Elizaveta Morozova
Source: Autism Speaks
The word "mitochondrion" has not yet entered every home, but in vain. Many people are aware that mitochondria are the "powerhouses" of our cells. However, mitochondria do more than just produce energy. Our mitochondria are sensitive to the environment in which we live, and they adapt the chemical processes in our cells to changes in the environment - cold, heat, dryness, humidity, infection, radiation, day, night, winter, summer, work, rest , sleep, gluttony, hunger, fear, love. All this is reflected in our mitochondria so that they can change the metabolism and behavior of our cells accordingly. Mitochondria have memory. When you exercise, your mitochondria grow and get better at adapting to the exact physical load that the body endures. When you eat in a certain way, mitochondria begin to respond better to these foods. The health and healthy development of our brain and body depends on the health of our mitochondria. The air, food and drinks you consume every day are processed by our mitochondria. They change to literally allow us to think, move, laugh and play, grow up, start families, meet a dignified old age.
Many people are aware that mitochondria are the "powerhouses" of our cells. However, mitochondria do more than just produce energy. Our mitochondria are sensitive to the environment in which we live, and they adapt the chemical processes in our cells to changes in the environment - cold, heat, dryness, humidity, infection, radiation, day, night, winter, summer, work, rest , sleep, gluttony, hunger, fear, love. All this is reflected in our mitochondria so that they can change the metabolism and behavior of our cells accordingly. Mitochondria have memory. When you exercise, your mitochondria grow and get better at adapting to the exact physical load that the body endures. When you eat in a certain way, mitochondria begin to respond better to these foods. The health and healthy development of our brain and body depends on the health of our mitochondria. The air, food and drinks you consume every day are processed by our mitochondria. They change to literally allow us to think, move, laugh and play, grow up, start families, meet a dignified old age. nine0003
nine0003
Schoffner's article and immunomitochondrial biology
Over the past few years, the special role of mitochondria in the response to infections has come to the fore in research. A new area of research has emerged that I call immunomitochondrial biology (IMB). MDI is a ray of light at the intersection of such seemingly different areas as mitochondrial disease and autism.
In a hit article by Schoffner and colleagues (1), published in 2009year, investigated the role of mitochondrial dysfunction as a risk factor for autism regression in a subgroup of children with autism spectrum disorders (ASD). Approximately 4% (5 out of 120 = 4.2%) of children with ASD definitely have mitochondrial disease (2, 3). Schoffner and colleagues selected 28 children who had both autism and a mitochondrial respiratory DNA disease.
They found that 17 out of 28 children (17/28=61%) had previously experienced a neurodegenerative episode when they first appeared and developed ASD symptoms.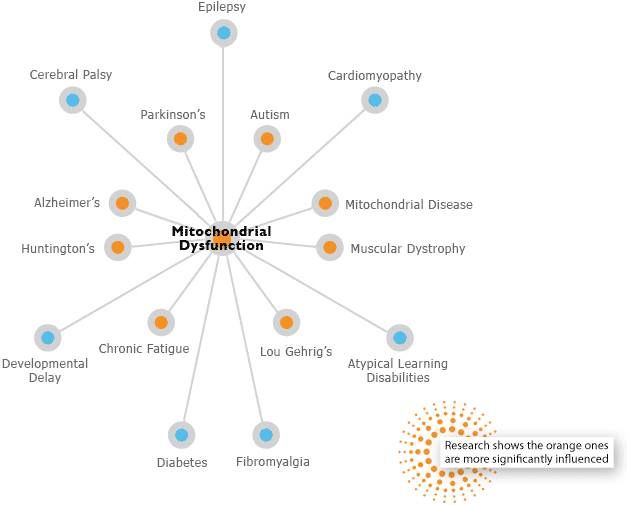 These are the so-called "autistic regressions", although it should be noted that the symptoms of ASD tend to develop and change over time after such a primary event (4). nine0003
These are the so-called "autistic regressions", although it should be noted that the symptoms of ASD tend to develop and change over time after such a primary event (4). nine0003
Typically, in autistic regressions, symptoms first appear within two weeks of an event such as an infectious disease, but they continue to develop and change over weeks to months after the acute reaction (4).
In 12 children (12/17 = 71%), regression began within two weeks after the increase in body temperature to 38.33 ° C. In 4 cases out of 12 (33% or 4/28 = 14% of all cases of ASD and mitochondrial disease) fever was caused by a reaction to standard vaccination. In 8 cases out of 12 (67%), the increase in body temperature was caused by an infection of unknown origin. nine0003
In patients with mitochondrial disease, fever may be due to some kind of infection or inflammation, or it may be "fever of unknown origin" when no cause has been identified. Five of the 17 patients (5/17=29%) had neither fever nor any documented infection. In 11 children out of 28 (11/28=39%), ASD developed gradually and did not have a single episode of regression in history.
In 11 children out of 28 (11/28=39%), ASD developed gradually and did not have a single episode of regression in history.
Neurodegeneration and mitochondrial disease
It is known that children with mitochondrial disease are at risk of degeneration of the nervous tissue after suffering an infectious disease (5). The risk of neurodegeneration (regression) after infection in the presence of mitochondrial disease without ASD was first documented by our research team at the University of California San Diego (5).
Robert Navier , MD, is an expert on mitochondrial DNA replication, DNA damage, signal nucleotide development, and their association with aging, healing, and regeneration. Author of several studies on the complex etiology of disorders such as diabetes and autism. He was the first to quantify the risk of neurodegenerative diseases in the case of a combination of infection and mitochondrial disease. His team was also able to demonstrate for the first time how defects in human DNA polymerase (mitochondrial DNA polymerase) can lead to disease. Co-inventor of several methods for the analysis of mitochondrial DNA. His main areas of research now are the development of new approaches to the treatment of diabetes and autism, as well as cancer and viral diseases, taking into account the intersections of innate immunity, inflammation, cellular genetics and metabolism. nine0003
Co-inventor of several methods for the analysis of mitochondrial DNA. His main areas of research now are the development of new approaches to the treatment of diabetes and autism, as well as cancer and viral diseases, taking into account the intersections of innate immunity, inflammation, cellular genetics and metabolism. nine0003
Interestingly, we reported exactly the same risk that was found by Schoffner and colleagues in 2009. In 2002, we reported that 60% of children with mitochondrial disease (18 out of 30 = 60%) suffered from neurodegenerative episodes (regressions), and 72% of these episodes (13/18 = 72%) were associated with infectious diseases during the period within two weeks before the start of the regression.
We did not find any difference between the number of infectious diseases that each child had during the year, which was 3-10 infectious diseases per year, which corresponds to the first few years of life of normal children (6). However, unlike healthy children, children with mitochondrial diseases were at risk of life-threatening neurodegeneration that followed the most common "childhood" diseases. In our study, regressions in classic mitochondrial disease did not lead to autism. However, such regressions resulted in severe loss of motor, language, or cognitive skills, and in some cases these losses were permanent. nine0003
In our study, regressions in classic mitochondrial disease did not lead to autism. However, such regressions resulted in severe loss of motor, language, or cognitive skills, and in some cases these losses were permanent. nine0003
Flash and fade reactions
There are two types of regression that parents and clinicians can learn to recognize. The two types differ in timing and symptoms after the triggering event, and they differ in the problems that follow. For simplicity, I call them "fade" and "flash" reactions.
When children with common forms of mitochondrial disease suffer regression, it is almost always a "fading out" response. The reaction usually occurs 2-10 days after the body temperature drops to normal levels (5), the same period of time I found for the course of Reye's syndrome at 1980s (7). Typically, parents report that their child has recovered from a cold or flu, and then, suddenly, the child's consciousness seems to fade away. The child is as if half asleep, he may stop walking, stop talking, his muscles become tense or lose their tone, he may have epileptic seizures or an episode resembling a stroke.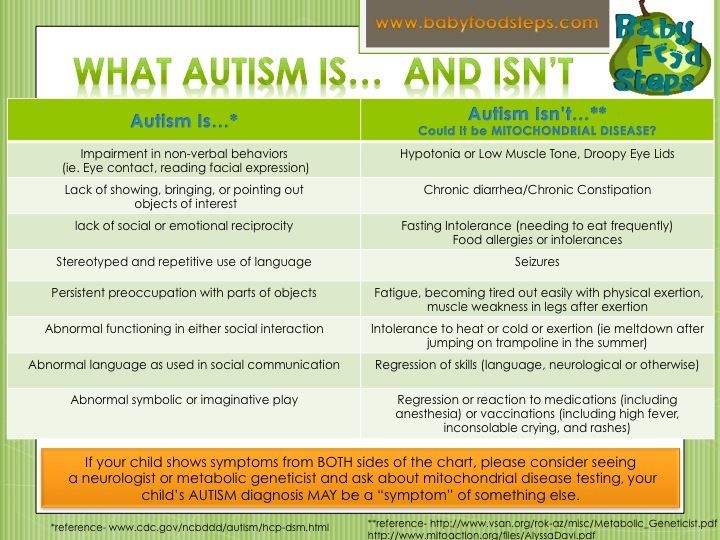 The extinction response is associated with a lack of energy, which can lead to a series of neurodegenerative episodes and even death over the next 2-3 months. In other cases, the child experiences only one episode, resembling a stroke, after which there is an improvement. nine0003
The extinction response is associated with a lack of energy, which can lead to a series of neurodegenerative episodes and even death over the next 2-3 months. In other cases, the child experiences only one episode, resembling a stroke, after which there is an improvement. nine0003
The process that leads to autistic regression proceeds quite differently. It usually refers to the 'outbreak' reaction and resembles the disease that Hannah Pauling suffered from and has been described in the scientific literature (4). An outbreak usually occurs early, during the peak of the fever and inflammatory response, within the first 2-3 days of infection. During the outbreak, there is a high temperature, often above 38.5 ° C, and is accompanied by excessive irritability, inconsolable crying, sleep disturbance, refusal to walk, when children who physically seem able to walk begin to crawl (4). After the flare reaction, a gradual development of problems begins, which is accompanied by severe digestive problems and diarrhea, a gradual loss of speech over 2-3 months, the appearance of repetitive movements, avoidance of eye contact and social contact (4).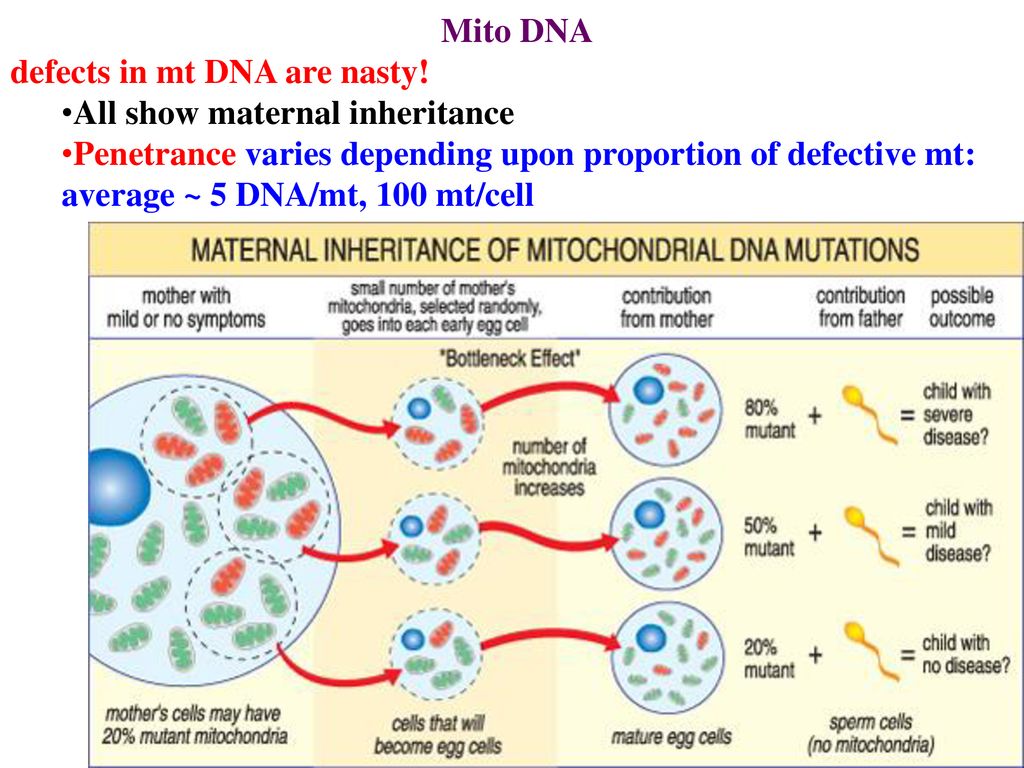 It must be emphasized that the flare reaction does not simply include a fever or even a severe reaction to a fever, such as a febrile convulsion. This is a multisystem inflammatory response that is associated with a risk of autistic regression for those children who have a genetic predisposition to it. One type of such a genetic predisposition is a mitochondrial disease. nine0003
It must be emphasized that the flare reaction does not simply include a fever or even a severe reaction to a fever, such as a febrile convulsion. This is a multisystem inflammatory response that is associated with a risk of autistic regression for those children who have a genetic predisposition to it. One type of such a genetic predisposition is a mitochondrial disease. nine0003
Different types of mitochondrial diseases
There are over 300 different types of mitochondrial diseases (8). Are there specific types of mitochondrial diseases that are associated with the risk of autistic regression, while other types are not associated with this risk? I think the answer is yes. In 2000, we reported a case of mitochondrial DNA (mtDNA) mutation (9) which at high concentration (86% heteroplasm) resulted in a severe form of classic mitochondrial disease in the older sister (Leigh syndrome), but at a lower concentration (61% heteroplasm). ) led to autism in a younger brother. nine0003
Although both children had the same mitochondrial DNA mutation, we found them to have completely different diseases and their development was excellent. There was no genotype-phenotype correlation. The results of an MRI scan of the brain of a sister with Leigh syndrome were very abnormal. The results of the magnetic resonance scan of the brother's brain were completely normal. The sister had delayed development and gradual development of problems, which was accompanied by a high concentration of lactic acid in the blood, seizures, cardiac anomalies, ataxia and involuntary movements. Despite this, her speech skills were almost normal. She did not have episodes of significant regression, and she did not lose skills after acquiring them, such cases were observed later. nine0003
There was no genotype-phenotype correlation. The results of an MRI scan of the brain of a sister with Leigh syndrome were very abnormal. The results of the magnetic resonance scan of the brother's brain were completely normal. The sister had delayed development and gradual development of problems, which was accompanied by a high concentration of lactic acid in the blood, seizures, cardiac anomalies, ataxia and involuntary movements. Despite this, her speech skills were almost normal. She did not have episodes of significant regression, and she did not lose skills after acquiring them, such cases were observed later. nine0003
In contrast, her brother was hyperactive, learned to use a few words normally, but lost them all at the age of two. He had a significant autistic regression without any known cause. The results of his blood and cerebrospinal fluid tests were normal. These facts should remind us that mitochondrial diseases, autism, and ASD are all neurological developmental disorders . The same factor in the lives of children with the same DNA mutations, but at different times in their lives, can lead to completely different consequences. nine0003
The same factor in the lives of children with the same DNA mutations, but at different times in their lives, can lead to completely different consequences. nine0003
Future research
We do not yet know how to determine which children are at such risk, but current scientific evidence suggests that children with milder and rarer forms of mitochondrial disease may be at higher risk of autistic regression than those who have severe forms of mitochondrial diseases that lead to multisystem disorders. Schoffner's scientific article, which has become a "hit", raises many important and new questions before us. nine0003
Can various mitochondrial defects cause fever and not vice versa? What specific mitochondrial defects lead to a rapidly developing and acute fever in response to an infectious disease or vaccination? What defects lead to an abnormal response to fever, or to a slight fever, or to a reduced response of the immune system to vaccination? Further research and careful observation by parents and pediatricians will help answer these questions, which will hopefully lead us to improved and encouraging treatments for children affected by these complex diseases. nine0003
nine0003
References
1. Shoffner, J., et al. Fever Plus Mitochondrial Disease Could Be Risk Factors for Autistic Regression. J Child Neurol (2009).
2. Oliveira, G., et al. Epidemiology of autism spectrum disorder in Portugal: prevalence, clinical characterization, and medical conditions. Dev Med Child Neurol 49, 726-733 (2007).
3. Bernier, F.P., et al. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 59, 1406-1411 (2002). nine0003
4. Poling, J.S., Frye, R.E., Shoffner, J. & Zimmerman, A.W. Developmental regression and mitochondrial dysfunction in a child with autism. J Child Neurol 21, 170-172 (2006).
5. Edmonds, J.L., et al. The otolaryngological manifestations of mitochondrial disease and the risk of neurodegeneration with infection. Arch Otolaryngol 128, 355-362 (2002).
6. Fox, J.P., Hall, C.E., Cooney, M.K., Luce, R.E. & Kronmal, R.A. The Seattle virus watch. II. Objectives, study population and its observation, data processing and summary of illnesses. Am J Epidemiol 96, 270-285 (1972).
Am J Epidemiol 96, 270-285 (1972).
7. Partin, J.C. Reye's Syndrome. in Liver Disease in Children (ed. Suchy, F.) 653-671 (Mosby, St. Louis, 1994).
8. Naviaux, R.K. Developing a systematic approach to the diagnosis and classification of mitochondrial disease. Mitochondrion 4, 351-361 (2004).
9. Graf, W.D., et al. Autism associated with the mitochondrial DNA G8363A transfer RNA(Lys) mutation. J Child Neurol 15, 357-361 (2000).
10. Prevalence of autism spectrum disorders – Autism and Developmental Disabilities Monitoring Network, United States, 2006. MMWR Surveill Summ 58, 1-20 (2009).
Autism Biomedicine, Research
Modern methods for preventing the transmission of mitochondrial DNA mutations through assisted reproductive technologies
Heteroplasmic mutations is a term used to describe the mixture of two or more different mitochondrial genomes in a single cell. The ratio of the amount of wild-type mitochondrial DNA (mtDNA) to the amount of mutant is called the degree of heteroplasmy and determines the severity of the disease.
mtDNA donor - a patient or individual (animal) with wild-type mtDNA.
Karyoplast - cell division spindle or nucleus, along with a small amount of cytoplasm, enclosed in a cell membrane; formed to transfer the nuclear genome of the recipient during enucleation.
Mitochondrial transfer - mutant mitochondria and, as a result, mtDNA, which are transferred along with the karyoplast during nuclear genome transfer (mitochondrial disease therapy). The amount of mtDNA is a function of the amount of cytoplasm and the density of mitochondria in the spindle or pronuclear region. nine0003
mtDNA Mismatch is an abnormal interaction between donor mitochondrial DNA and recipient mtDNA after nuclear genome transfer due to differences in DNA sequences. Occurs as a result of mitochondrial transfer.
mtDNA recipient - a patient or individual (animal) with mutant mtDNA and signs of disease.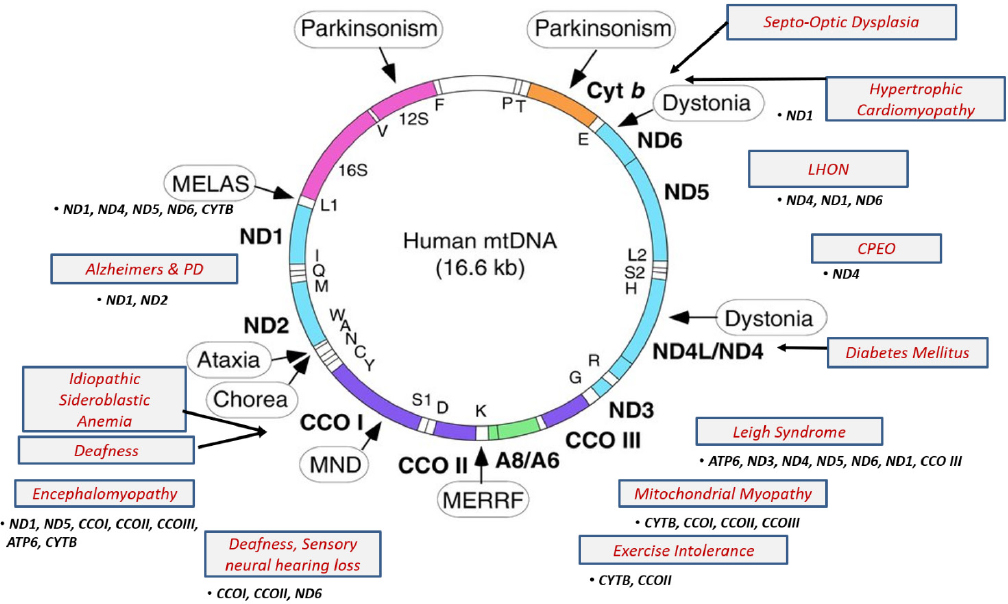
Cytoplast - a cell membrane with cytoplasm, excluding the nucleus, formed after cell enucleation or spindle removal procedure. nine0003
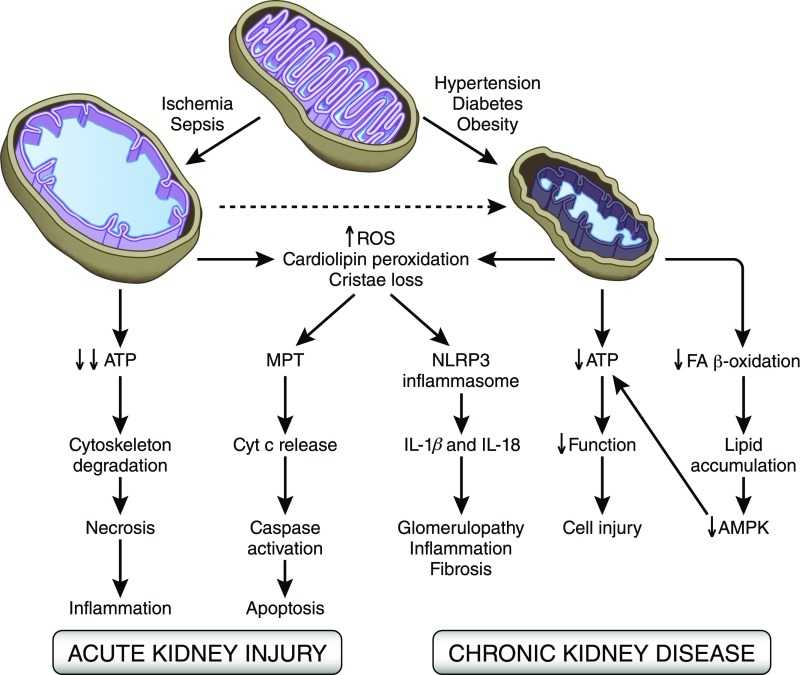
Defects in the functional state of mitochondria are increasingly discussed in the literature [1], being the cause of many serious diseases affecting various organs and tissues. Mitochondria are under the genetic control of both the nuclear and mitochondrial genomes, defects in one of which result in mitochondrial dysfunction. Many adults and children with mitochondrial diseases carry inherited defects in the mitochondrial genome, with at least 1 in 8500 individuals in the population carrying mtDNA. This means that at least 1/3 of women of reproductive age are carriers of mtDNA mutations [2]. nine0003
mtDNA defects were first described in 1988. Since then, methods for diagnosing mitochondrial diseases have improved, but there are still many limitations in the treatment of these disorders [3, 4]. mtDNA encodes 13 main subunits of the respiratory chain, as well as 24 RNA molecules required for intramitochondrial protein synthesis [4]. The mitochondrial genome is present in multiple copies in a cell. In healthy individuals, all its copies are identical - homoplasmic [1, 4]. In patients with genetic defects, mutations may be present in all copies of the genome (homoplasmic mutations) or only in some copies (heteroplasmic mutations). Due to the difficulties in the treatment of mitochondrial diseases, the priority is to provide families with appropriate genetic counseling, as well as to develop ways to prevent the transmission of these diseases [1, 5]. nine0003
The mitochondrial genome is present in multiple copies in a cell. In healthy individuals, all its copies are identical - homoplasmic [1, 4]. In patients with genetic defects, mutations may be present in all copies of the genome (homoplasmic mutations) or only in some copies (heteroplasmic mutations). Due to the difficulties in the treatment of mitochondrial diseases, the priority is to provide families with appropriate genetic counseling, as well as to develop ways to prevent the transmission of these diseases [1, 5]. nine0003
Clinically, mitochondrial diseases are very diverse and occur in patients of all age groups. Patients turn to doctors of a wide range of specialties. mtDNA mutations are associated with serious disorders such as myopathies, neurodegenerative diseases, hematopoietic diseases, diabetes mellitus, cancer, and infertility [3, 5]. Examples of such diseases are: progressive ophthalmoplegia, Pearson's syndrome (pancytopenia, insulin-dependent diabetes mellitus), Leber's optic neuropathy, Leigh's syndrome (subacute necrotizing encephalopathy), various types of myodystrophy and myosthenia, acute skeletal muscle necrosis (rhabdomyolysis), as well as aminoglycoside-induced hearing loss and other neurodegenerative diseases.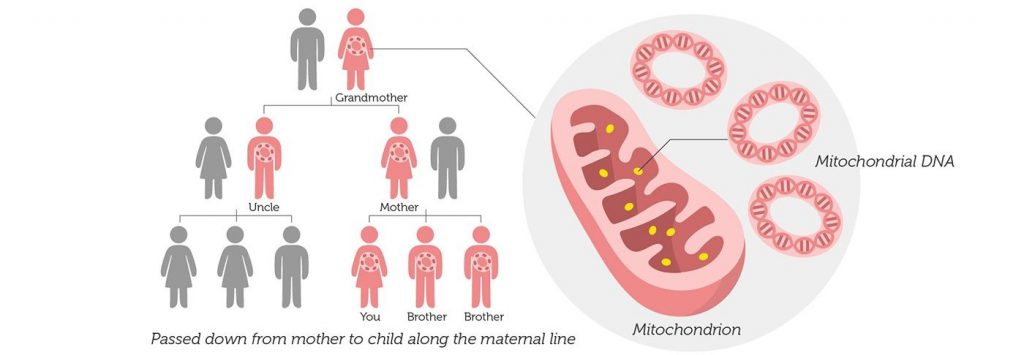 In addition, animal and human studies show that mtDNA mutations and mitochondrial dysfunction are involved in the pathogenesis of age-related changes and diseases such as diabetes mellitus, Alzheimer's disease, Parkinson's and Huntington's diseases [5]. nine0003
In addition, animal and human studies show that mtDNA mutations and mitochondrial dysfunction are involved in the pathogenesis of age-related changes and diseases such as diabetes mellitus, Alzheimer's disease, Parkinson's and Huntington's diseases [5]. nine0003
This literature review highlights aspects of current clinical practice and future opportunities within ART to prevent the transmission of mtDNA mutations. Including the use of new technologies for micromanipulation of zygote pronuclei, transfer of the fission spindle and polar body between embryos: modern approaches that have been shown to be successfully implemented in animals.
Genetic counseling nine0002 mtDNA is maternally inherited. Family studies have shown that mutations can occur spontaneously or can be inherited, affecting many family members [1, 6]. Mutations vary widely in transmission risks, for example, single mtDNA deletions are often sporadic, while others are transmitted with a high degree of probability [5].
 Identification of specific mutations and examination of all family members can be useful in determining the probability of transmission of the disease to offspring [7]. Detailed studies in large cohorts of patients also provide valuable information about the risk of transmission. For example, an analysis of 226 families in which deletions in the mtDNA genes were identified in the proband showed that the risk of recurrence in the offspring of a sick mother is 4-11%. If the mother has no clinical symptoms, and there is no information about the affected brothers and sisters of the proband, it is assumed that the risk of relapse is low [1, 8]. nine0003
Identification of specific mutations and examination of all family members can be useful in determining the probability of transmission of the disease to offspring [7]. Detailed studies in large cohorts of patients also provide valuable information about the risk of transmission. For example, an analysis of 226 families in which deletions in the mtDNA genes were identified in the proband showed that the risk of recurrence in the offspring of a sick mother is 4-11%. If the mother has no clinical symptoms, and there is no information about the affected brothers and sisters of the proband, it is assumed that the risk of relapse is low [1, 8]. nine0003 Maternal transmission of mtDNA mutations is well described and defined [5], although the outcome of specific pregnancies remains unpredictable, especially for heteroplasmic disorders. This is largely due to genetic elimination occurring during early development [1, 3], which leads to significant changes in DNA and mutant load in the offspring [5]. Many of the clinical symptoms correlate with the ratio of the amount of mutant mtDNA to the amount of wild-type mtDNA (or the absolute amount of wild-type DNA), so the outcome for each pregnancy is difficult to predict [1, 4, 5, 9, 10].
Many of the clinical symptoms correlate with the ratio of the amount of mutant mtDNA to the amount of wild-type mtDNA (or the absolute amount of wild-type DNA), so the outcome for each pregnancy is difficult to predict [1, 4, 5, 9, 10].
In the case of homoplasmic mtDNA disorders, the mutation is transmitted to all offspring [1, 5]. However, even in such patients, the development of clinical signs is difficult to predict, since there is a variable penetrance, apparently due to a combination of environmental factors and genetic control of the nucleus [5]. Thus, genetic counseling for families with mitochondrial diseases remains a challenge [1, 9, 10].
Egg donation
Donor eggs from an unrelated person are an option to prevent the transmission of mtDNA mutations to offspring. Nevertheless, despite the fact that fertilization is carried out with the help of the father's sperm, the child will have the donor's nuclear genotype and genetic relationship only with the father [1]. Some women who need donor eggs express concern about this and experience severe psychological discomfort, up to depression symptoms [11]. In addition, in many countries there are various difficulties with obtaining donor eggs and choosing an oocyte donor. Another limitation is that this procedure often takes more time than a standard ART program with own oocytes, largely due to the donor waiting time [1, 11]. nine0003
Some women who need donor eggs express concern about this and experience severe psychological discomfort, up to depression symptoms [11]. In addition, in many countries there are various difficulties with obtaining donor eggs and choosing an oocyte donor. Another limitation is that this procedure often takes more time than a standard ART program with own oocytes, largely due to the donor waiting time [1, 11]. nine0003
Chorionic biopsy and amniocentesis
Amniocentesis and chorionic villus sampling are widely used to diagnose chromosomal abnormalities and can be used to diagnose mtDNA mutations. When these methods were used to diagnose specific mtDNA mutations (8993T>G, 8993T>C), the results influenced pregnancy management [1, 7]. In the case of heteroplasmic mtDNA disorders, tissue-specific differences in the level of heteroplasmy are often noted, making it doubtful that this test will accurately reflect fetal outcome. However, existing data suggest that in most cases this is not a problem [1, 5].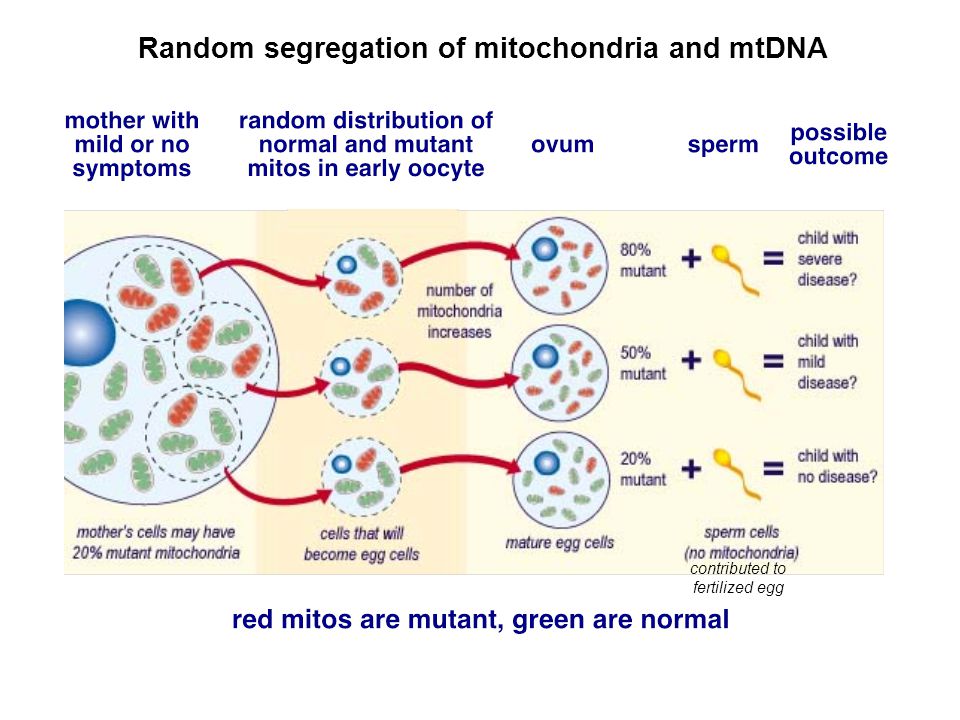 Using this test, it is only possible to predict the outcome for a very high or very low level of heteroplasmy and only for some mutations. Prediction is difficult for intermediate values [7, 12]. nine0003
Using this test, it is only possible to predict the outcome for a very high or very low level of heteroplasmy and only for some mutations. Prediction is difficult for intermediate values [7, 12]. nine0003
Preimplantation genetic diagnosis
Preimplantation genetic diagnosis (PGD) allows analyzing mtDNA from the polar bodies of unfertilized oocytes, individual blastomeres of 6-8 cell embryos or trophectoderm cells with subsequent transfer of only healthy embryos into the uterine cavity. Currently, PGD is the method of choice for patients with hereditary mtDNA defects [6, 13]. It has been shown that each cell has, on average, about 10 to 100,000 mtDNA copies [13]. Although copy number may vary between cells of different embryos and different stages of development, the high copy number suggests that it is possible to conduct a PGD program for mitochondrial diseases. There is a concern that polar bodies or individual cells cannot reflect the real amount of DNA from the total mutation load, although experiments on heteroplasmic mice and human blastomeres have shown a homogeneous distribution at the beginning of embryogenesis [1, 13]. nine0003
nine0003
Development of new methods
Genetic counseling, PGD, and prenatal diagnosis may be useful for some patients with heteroplasmic mtDNA mutations, but these methods are ineffective in treating patients with homoplasmic mutations. For these families, it is important to develop alternative methods to prevent the transmission of mtDNA diseases [1, 5, 13, 14].
Cytoplasmic transfer
In this technique, normal cytoplast mitochondria of the donor are transferred to the recipient oocyte and thus dilute the effects of any mtDNA defect [15]. Cytoplasmic transfer between human oocytes was performed for the first time to try to improve the results of ART [12, 15]. However, some of the children born were heteroplasmic with a low level of mtDNA from oocyte donors [1, 14]. Therefore, this method is likely to be of little value in patients with mitochondrial dysfunction. Experiments on mice show that the amount of donor mtDNA during cytoplasmic transfer is less than 1/3 of the total amount of mtDNA, so the proportion of wild-type mtDNA will change little. In addition, there are concerns that cytoplasm transfer can cause serious epigenetic modifications [12], as well as chromosomal abnormalities [1, 14]. nine0003
Experiments on mice show that the amount of donor mtDNA during cytoplasmic transfer is less than 1/3 of the total amount of mtDNA, so the proportion of wild-type mtDNA will change little. In addition, there are concerns that cytoplasm transfer can cause serious epigenetic modifications [12], as well as chromosomal abnormalities [1, 14]. nine0003
Nuclear genome transfer into enucleated donor oocytes
An alternative strategy involves the transfer of nuclear DNA from a mother with mtDNA disease to enucleated oocytes or embryos from a healthy female oocyte donor [12, 16, 17]. There are some difficulties associated with these procedures. In the metaphase II stage, the oocyte does not have a nuclear membrane, so there is a high risk of losing chromosomes during the procedure. The use of fluorophores to visualize chromosomes in this case may affect further normal embryonic development. If oocytes are used in the germinal vesicle (GV) stage, the viability of the resulting embryos is significantly reduced in vitro , which was demonstrated in human and mouse oocytes [1, 8, 16, 17]. Nevertheless, modern approaches to the transfer of the nuclear genome into enucleated oocytes involve the use of sufficiently effective methods; protocols for the procedures have been published and are available [17-21]. This strategy can be implemented by several methods [17, 21] described below.
Nevertheless, modern approaches to the transfer of the nuclear genome into enucleated oocytes involve the use of sufficiently effective methods; protocols for the procedures have been published and are available [17-21]. This strategy can be implemented by several methods [17, 21] described below.
Transfer of zygote pronuclei nine0002 The zygote stage in mammals is characterized by the presence of two pronuclei - male and female, each is clearly visualized and contains a haploid set of nuclear (genomic) DNA chromosomes. A schematic representation of the transfer of zygote pronuclei is shown in Fig. 1 [17, 21]. The transfer of pronuclei between zygotes was first carried out in early 1980 in mice, demonstrating that manipulations with mouse zygotes can produce viable offspring [22]. More recently, pronuclear transfer between zygotes has been used to model the prevention of transmission of mitochondrial diseases [16]. The efficiency of zygote pronucleus transfer in mice is characterized by high mitochondrial transfer in offspring and remains low in 24% of cases [17, 23].
 This is due to the inevitable co-transfer of a small amount of cytoplasm containing mitochondria and mutant mtDNA (see Fig. 1). Thus, the isolation of pronuclei, even in small karyoplasts, can lead to the joint transfer of a large number of mitochondria [17]. Although most common hereditary human mtDNA diseases are usually associated with high levels of mutant mtDNA [5], these results in mice do not herald the widespread use of this method for the treatment of mitochondrial disorders in human zygotes [8, 17]. Nevertheless, the possibility of pronucleus transfer between human zygotes was first demonstrated in 2010 [24]. L. Craven et al. [17, 24] studied abnormal zygotes containing one or more than two pronuclei, which are usually utilized routinely in ART cycles. Zygotes containing two pronuclei were reconstructed by transferring one pronucleus from a polypronuclear zygote to haploid zygotes. Only half of the reconstructed zygotes contained both male and female pronucleus due to the difficulty of differentiating them from each other during the procedure.
This is due to the inevitable co-transfer of a small amount of cytoplasm containing mitochondria and mutant mtDNA (see Fig. 1). Thus, the isolation of pronuclei, even in small karyoplasts, can lead to the joint transfer of a large number of mitochondria [17]. Although most common hereditary human mtDNA diseases are usually associated with high levels of mutant mtDNA [5], these results in mice do not herald the widespread use of this method for the treatment of mitochondrial disorders in human zygotes [8, 17]. Nevertheless, the possibility of pronucleus transfer between human zygotes was first demonstrated in 2010 [24]. L. Craven et al. [17, 24] studied abnormal zygotes containing one or more than two pronuclei, which are usually utilized routinely in ART cycles. Zygotes containing two pronuclei were reconstructed by transferring one pronucleus from a polypronuclear zygote to haploid zygotes. Only half of the reconstructed zygotes contained both male and female pronucleus due to the difficulty of differentiating them from each other during the procedure. Reconstructed zygotes ( n =36) were cultured to the blastocyst stage. Three zygotes, or 8%, reached the blastocyst stage, mtDNA analysis showed a low percentage (less than 2%) of mitochondrial transfer. Despite the low percentage of obtained blastocysts, the authors [24] concluded that the transfer of zygote pronuclei is promising for the treatment of mitochondrial diseases in humans. However, it is currently not possible to evaluate the safety and efficacy of zygote pronuclear transfer in normal human embryos. Based on this study, further study of the method is required for the final conclusion on the justification for the use of pronuclear transfer for the treatment of mitochondrial diseases in humans [3, 17, 21]. nine0003
Reconstructed zygotes ( n =36) were cultured to the blastocyst stage. Three zygotes, or 8%, reached the blastocyst stage, mtDNA analysis showed a low percentage (less than 2%) of mitochondrial transfer. Despite the low percentage of obtained blastocysts, the authors [24] concluded that the transfer of zygote pronuclei is promising for the treatment of mitochondrial diseases in humans. However, it is currently not possible to evaluate the safety and efficacy of zygote pronuclear transfer in normal human embryos. Based on this study, further study of the method is required for the final conclusion on the justification for the use of pronuclear transfer for the treatment of mitochondrial diseases in humans [3, 17, 21]. nine0003 Rice. 1. Schematic representation of the zygote pronuclear transfer procedure. Male and female pronuclei are removed from donor and patient zygotes with the formation of a donor cytoplast and a karyoplast with the patient's nuclear genome using special microtools.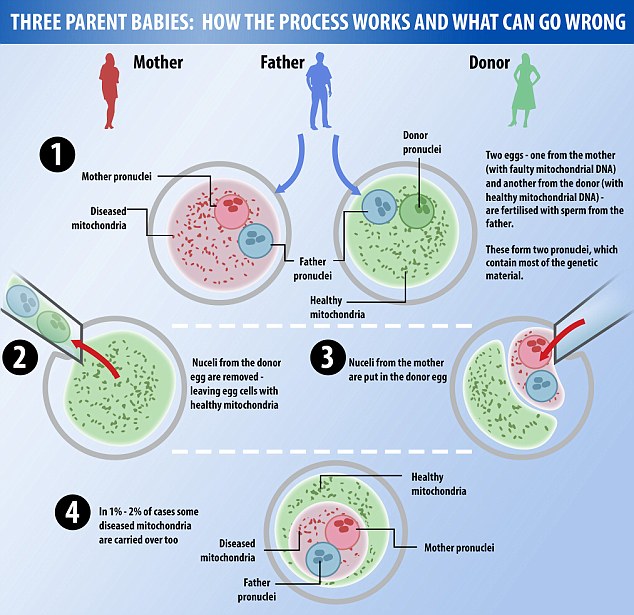 After binding of the cytoplast and karyoplast, the embryos are ready for cultivation and further transfer. The patient's karyoplast contains mutant mitochondria that are transferred into the reconstructed embryo. The reconstructed zygote may still contain unacceptable amounts of mutant mtDNA, the so-called mitochondrial transfer. Reproduced and modified accordingly [17]. nine0003
After binding of the cytoplast and karyoplast, the embryos are ready for cultivation and further transfer. The patient's karyoplast contains mutant mitochondria that are transferred into the reconstructed embryo. The reconstructed zygote may still contain unacceptable amounts of mutant mtDNA, the so-called mitochondrial transfer. Reproduced and modified accordingly [17]. nine0003
Spindle transfer
In 2009, spindle transfer was performed for the first time in an animal model (rhesus monkey), demonstrating that high efficiency of the nuclear genome transfer technique can be achieved at the stage of unfertilized oocytes [2]. These experiments demonstrated the efficiency and safety of spindle transfer: viable offspring were obtained that developed normally (growth curves did not differ from those of the control group), moreover, the level of mitochondrial transfer was low (below 2%) [2, 17-21 , 25]. A technical description of the spindle transfer protocol has been published [17–19] and is shown in fig. 2. Unlike zygotes, the distribution of mitochondria in oocytes is homogeneous, which allows the transfer of the fission spindle without significant mitochondrial transfer of mutant mtDNA. Karyoplasts with a volume of about 1.5% of the volume of the donor cytoplast contained a meiotic spindle with a small volume of cytoplasm. After oocyte reconstruction, fertilization was performed by intracytoplasmic sperm injection (ICSI). The resulting embryos were cultured to the blastocyst stage. The quality and development of blastocysts were comparable to the control group [2]. Fifteen blastocysts transferred 9surrogate mothers, resulting in three pregnancies and 4 live births. The results were similar to those with non-reconstructed embryos [2, 17, 20].
2. Unlike zygotes, the distribution of mitochondria in oocytes is homogeneous, which allows the transfer of the fission spindle without significant mitochondrial transfer of mutant mtDNA. Karyoplasts with a volume of about 1.5% of the volume of the donor cytoplast contained a meiotic spindle with a small volume of cytoplasm. After oocyte reconstruction, fertilization was performed by intracytoplasmic sperm injection (ICSI). The resulting embryos were cultured to the blastocyst stage. The quality and development of blastocysts were comparable to the control group [2]. Fifteen blastocysts transferred 9surrogate mothers, resulting in three pregnancies and 4 live births. The results were similar to those with non-reconstructed embryos [2, 17, 20].
Rice. 2. Schematic representation of the procedure for transferring the fission spindle. The spindle is visualized using a special microscope optical system and removed from MII oocytes of the donor and the patient. With the help of special microtools, a donor cytoplast and a karyoplast with the patient's nuclear genome are formed.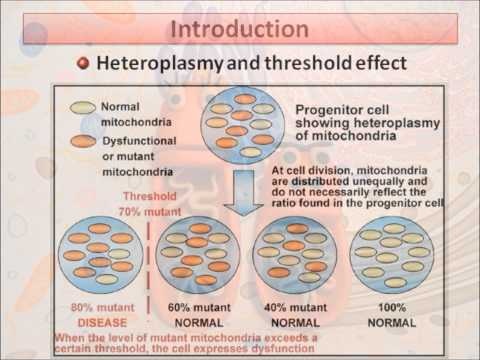 After the binding of the cytoplast and karyoplast, a reconstructed oocyte is formed, which is fertilized with the partner's sperm. The patient's karyoplast contains mutant mitochondria (less than 2%), which are transferred to the reconstructed oocyte. Reproduced and modified accordingly [17]. nine0003
After the binding of the cytoplast and karyoplast, a reconstructed oocyte is formed, which is fertilized with the partner's sperm. The patient's karyoplast contains mutant mitochondria (less than 2%), which are transferred to the reconstructed oocyte. Reproduced and modified accordingly [17]. nine0003
Studies on human oocytes from donors with different mtDNA haplotypes were carried out later [25]. Of the 106 eggs donated for the study, 65 underwent spindle transfer and 33 constituted the control group. The frequency of fertilization in the experimental group was 73% and was close to the control (75%). However, a significant proportion (52%) of zygotes after spindle transfer demonstrated abnormal fertilization (more often, the presence of three pronuclei) [17, 21, 25]. The level of heteroplasmy in the resulting embryos was less than 1%. The results of this study are consistent with the conclusion that spindle transfer can be an effective and safe treatment for mitochondrial diseases with minimal mitochondrial transfer [2, 3, 17]. nine0003
nine0003
Transport of polar bodies
During meiosis, mammalian oocytes undergo two reductive divisions with unequal cytoplasmic separation and the formation of two small bodies containing an additional set of chromosomes. The first polar body contains a diploid set of chromosomes, and the second a haploid set. Completion of meiosis and isolation of the first and second polar bodies is necessary for proper normal embryonic development [23, 26]. Polar body transfer in the context of therapy for mitochondrial diseases has been described in the literature [21, 23] (Fig. 3). The basis of polar body transfer for the treatment of mitochondrial disorders is that polar bodies contain genomic DNA and only a few mitochondria. In addition, polar bodies are easily visualized and can be transferred to donor cytoplasts [17]. Using this approach on mouse oocytes, the first polar body and spindle were simultaneously transferred (see Fig. 3), 43 reconstructed oocytes were obtained. The birth rate in the experimental group was comparable to the control group, and all live puppies obtained from reconstructed eggs developed normally [17, 23, 26]. However, the transfer of the polar body did not give results in experiments on other mammals, despite significant efforts [3, 12, 17]. In most species, after the transfer of the polar body, low embryonic viability, DNA fragmentation and degeneration were observed. In clinical practice, human polar bodies are of great importance for PGD of some diseases, however, the efficacy and safety of polar body transfer for the treatment of mitochondrial disorders remains to be determined. It should be noted that due to recombination processes in meiosis, the genetic composition of the polar bodies is not identical to the genetic composition of the oocyte or zygote, which is probably why the transfer of the polar body is ineffective [3, 17]. nine0003
The birth rate in the experimental group was comparable to the control group, and all live puppies obtained from reconstructed eggs developed normally [17, 23, 26]. However, the transfer of the polar body did not give results in experiments on other mammals, despite significant efforts [3, 12, 17]. In most species, after the transfer of the polar body, low embryonic viability, DNA fragmentation and degeneration were observed. In clinical practice, human polar bodies are of great importance for PGD of some diseases, however, the efficacy and safety of polar body transfer for the treatment of mitochondrial disorders remains to be determined. It should be noted that due to recombination processes in meiosis, the genetic composition of the polar bodies is not identical to the genetic composition of the oocyte or zygote, which is probably why the transfer of the polar body is ineffective [3, 17]. nine0003
Rice. 3. Schematic representation of the transfer procedure of the first polar body together with the transfer of the fission spindle. The spindle is visualized and removed from the donor oocyte to form the donor cytoplast. A karyoplast with the patient's nuclear genome is formed from the first polar body using special microtools. After binding of the cytoplast and karyoplast, the reconstructed oocyte is fertilized with partner's sperm. The patient's karyoplast contains only a few mitochondria, which are transferred to the reconstructed oocyte. Reproduced and modified according to [17]. nine0003
The spindle is visualized and removed from the donor oocyte to form the donor cytoplast. A karyoplast with the patient's nuclear genome is formed from the first polar body using special microtools. After binding of the cytoplast and karyoplast, the reconstructed oocyte is fertilized with partner's sperm. The patient's karyoplast contains only a few mitochondria, which are transferred to the reconstructed oocyte. Reproduced and modified according to [17]. nine0003
Genetic counseling for families with mitochondrial dysfunction is important because the risks of transmission differ for different mtDNA mutations. Physicians with experience in this field should ensure proper counseling and support for families with an identified risk of transmitting the disease to offspring [1, 7].
Currently, PGD is the method of choice for reducing the risk of transmission of mitochondrial disorders, especially in women with low levels of mtDNA heteroplasmy [13]. However, the successful use of PGD depends on the number of high quality embryos with a low level of mutant load, which makes it unsuitable for some patients [1, 13]. nine0003
nine0003
New methods of micromanipulation with unfertilized oocytes and zygotes, which are an alternative to donor eggs, are a promising and promising direction for the prevention of mtDNA diseases [3, 12, 17]. However, the safety and efficacy of these methods should be carefully studied in the future. None of the above methods guarantees the complete elimination of the patient's mutant mtDNA. Therefore, further studies on animal models are needed to prove that the penetrance of mtDNA heteroplasmy will not affect the viability of offspring with all methods of nuclear genome micromanipulation before these methods enter clinical practice [3, 8, 12]. Safety control should not be limited to the early stages of development, but should be extended to the observation of adult offspring, preferably in several generations [3]. Available data obtained on human donor oocytes refer only to the period of early embryonic development [25], these data are clearly not enough to consider these methods safe. Studies of early events by observing morphokinetics can be useful [3]. It is also important to study the chromosomal status of the obtained blastocysts, as well as transcriptome, proteome, and epigenome analysis data. Long-term analysis of subsequent generations in animal models is required to provide full empirical proof of the principle of the absence of side effects after mitochondrial replacement [3, 8, 12, 17, 21]. nine0003
Studies of early events by observing morphokinetics can be useful [3]. It is also important to study the chromosomal status of the obtained blastocysts, as well as transcriptome, proteome, and epigenome analysis data. Long-term analysis of subsequent generations in animal models is required to provide full empirical proof of the principle of the absence of side effects after mitochondrial replacement [3, 8, 12, 17, 21]. nine0003
Despite the technical and ethical problems of these approaches, mitochondrial donation holds great promise, allowing women carrying mtDNA mutations to have a genetically related child without passing on the mutant mtDNA, thereby preventing the transmission of mitochondrial disorders [11, 12].
Legislation regarding research using germ cells and embryos varies considerably from country to country. In the United Kingdom, an expert council based on the HFEA (Human Fertilization and Embryology Authority) has been created to review methods to prevent mitochondrial diseases.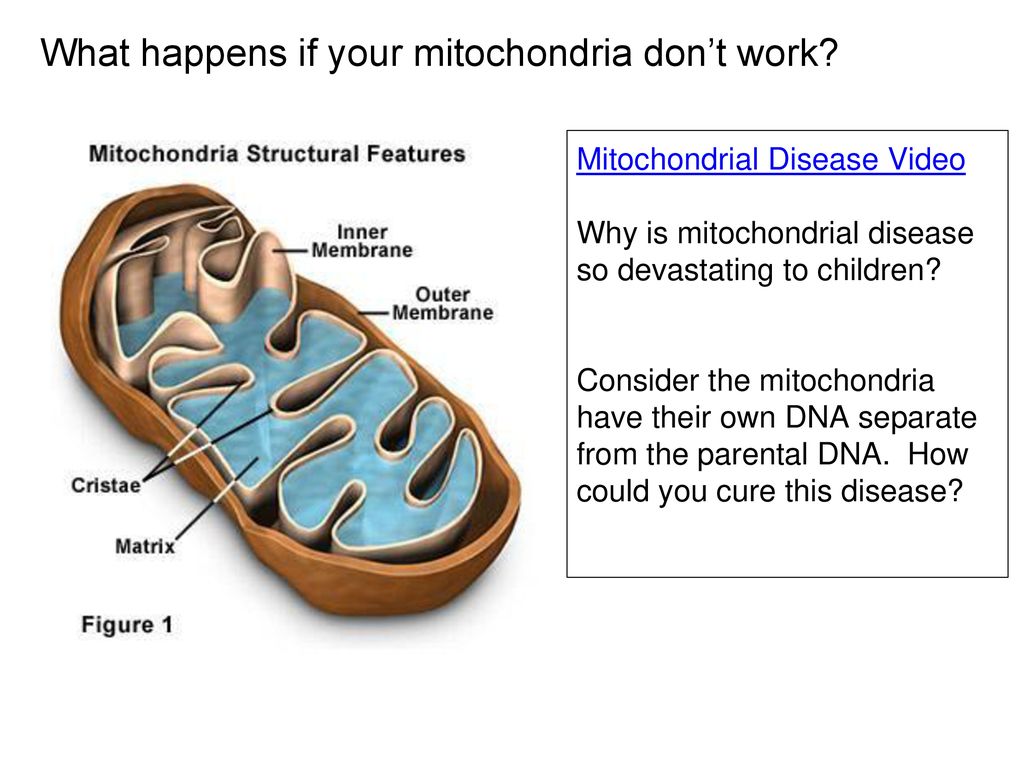 A report on the evaluation of the safety and efficacy of therapies for mitochondrial diseases can be found on the HFEA website (www.hfea.gov.uk/6372.html). HFEA has received wide public support for mitochondrial donation in the UK [3]. The expert panel concluded that zygote and spindle pronuclear transfer techniques have the potential to be of great benefit to patients with severe hereditary mtDNA diseases. The Expert Council strongly recommends further additional research to ensure the efficacy and safety of these methods prior to their clinical use. Long-term follow-up of children born as a result of these methods is also recommended [3, 12, 21]. In June 2013, the Government of the United Kingdom announced its decision to proceed with draft regulations that would allow the use of nuclear genome transfer and mitochondrial replacement techniques for the treatment of patients, and already in 2015 these methods were approved for therapeutic purposes by the House of Lords of the United Kingdom.
A report on the evaluation of the safety and efficacy of therapies for mitochondrial diseases can be found on the HFEA website (www.hfea.gov.uk/6372.html). HFEA has received wide public support for mitochondrial donation in the UK [3]. The expert panel concluded that zygote and spindle pronuclear transfer techniques have the potential to be of great benefit to patients with severe hereditary mtDNA diseases. The Expert Council strongly recommends further additional research to ensure the efficacy and safety of these methods prior to their clinical use. Long-term follow-up of children born as a result of these methods is also recommended [3, 12, 21]. In June 2013, the Government of the United Kingdom announced its decision to proceed with draft regulations that would allow the use of nuclear genome transfer and mitochondrial replacement techniques for the treatment of patients, and already in 2015 these methods were approved for therapeutic purposes by the House of Lords of the United Kingdom.