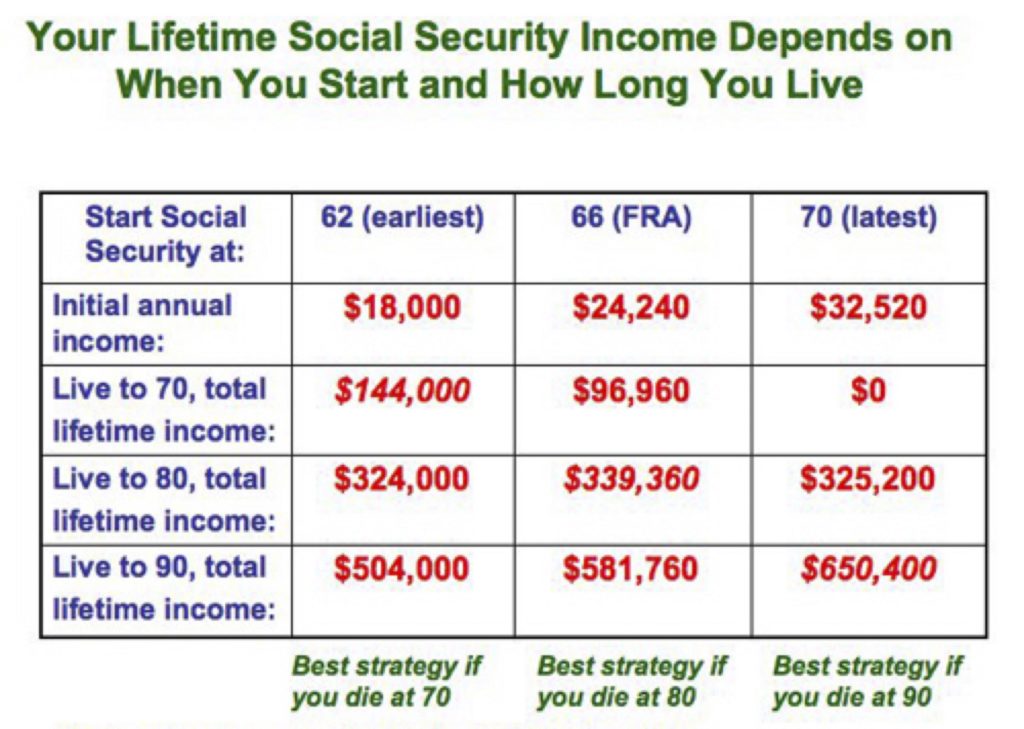
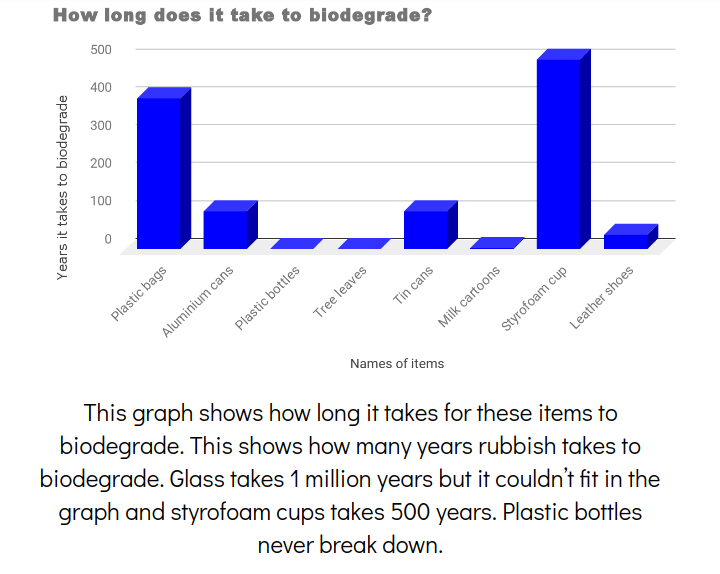
How long does ssi last for a child
Parents and Guardians | SSA
With you through life’s journey...
The makeup of American families has changed in the last 20 to 30 years. Today, family units are diverse, rich in culture, and may include two parents, same-sex parents, only one parent, grandparents, and other relatives. Social Security knows that whether single parent, blended, diverse, small or large, every family is important.
For more than 80 years, Social Security has helped families secure today and tomorrow by providing financial benefits, tools, and programs that help support millions throughout life’s journey. Our programs and services have evolved to meet your unique family needs and especially the children in your care.
We are there from day one
Getting your child a Social Security number should be near the top of the list of things you need to do as a new parent or guardian. Your child's Social Security number is the first step in ensuring valuable protection for any benefits they may be eligible for in the future.
You’ll need your child’s Social Security number to claim them as a dependent on your income tax return or open a bank account in the child’s name and buy savings bonds. Your child’s Social Security number is also necessary to obtain medical coverage or apply for any kind of government services for your child.
Most people apply for their child’s Social Security number at birth, usually at the hospital. When the time comes for your child’s first job, the number is already in place. For more information on getting your child a Social Security number and card, check out Social Security Numbers for Children.
A fun bonus of assigning Social Security numbers at birth is that we know the most popular baby names, which we announce each year. On our website, you can find the top baby names for the last 100 years.
We’re there with support if you’re raising a grandchild…
More and more grandparents are finding themselves raising their grandchildren. Social Security will pay benefits to grandchildren when the grandparent retires, becomes disabled, or dies, if certain conditions are met. Generally, the biological parents of the child must be deceased or disabled, or the grandparent must legally adopt the grandchild.
Social Security will pay benefits to grandchildren when the grandparent retires, becomes disabled, or dies, if certain conditions are met. Generally, the biological parents of the child must be deceased or disabled, or the grandparent must legally adopt the grandchild.
To receive this benefit, your grandchild must have begun living with you before age 18 and received at least one half of his or her support from you for the year before the month you became entitled to retirement or disability insurance benefits, or died. Also, the natural parent(s) of the child must not be making regular contributions to his or her support.
If your grandchild was born during the one-year period, you must have lived with and provided at least one-half of the child's support for substantially the entire period from the date of birth to the month you became entitled to benefits.
Your grandchild may qualify for benefits under these circumstances, even if he or she is a step-grandchild. However, if you and your spouse are already receiving benefits, you would need to adopt the child for them to qualify for benefits.
However, if you and your spouse are already receiving benefits, you would need to adopt the child for them to qualify for benefits.
We’re there when they get their first job
Once your child starts working and throughout their career, employers will verify their Social Security number to help reduce fraud and improve the accuracy of their earnings records.
Employers collect FICA, or Federal Insurance Contributions Act withholdings, and report earnings electronically. This is how we verify earnings and is how your child earns Social Security retirement, disability, and survivors coverage.
Once they turn 18, they can open a my Social Security account and watch their personal earnings and future benefits grow over time.
We’re there to help if disability strikes…
As a working parent, your earnings can become a source of Social Security protection for your family. If you retire or become disabled and unable to work, your earnings would be partially replaced by your monthly Social Security benefit payments.
A child who is disabled may depend on your help for a lifetime. When you start receiving Social Security retirement or disability benefits, your family members also may be eligible for payments. If you are a parent, caregiver, or representative of a child younger than age 18 who has a disability, your child may be eligible for Supplemental Security Income (SSI) payments. More information is provided in the Benefits for Children with Disabilities booklet.
For children 18 years or older who have been disabled before the age of 22 and continue to be disabled, Social Security benefits may be paid to them if you retire, become disabled, or die. Social Security benefits for disabled children may continue as long as they are unable to work because of their disability.
Additionally, you can find information on the specific benefits and qualifications in the Disability Benefits publication.
We’re there to provide comfort during difficult times…
The loss of a parent or guardian can be both emotionally and financially difficult. Social Security helps by providing benefits to help stabilize the family’s financial future. Widows, widowers, and their dependent children may be eligible for Social Security survivors benefits.
Social Security helps by providing benefits to help stabilize the family’s financial future. Widows, widowers, and their dependent children may be eligible for Social Security survivors benefits.
In fact, 98 of every 100 children could get benefits if a working parent dies. And Social Security pays more benefits to children than any other federal program.
Providing protection for parents too…
Even if you have never worked in a job covered by Social Security, as a parent, there are two ways that you may still qualify for benefits.
- If you are a parent and take care of your child who receives Social Security benefits and is under age 18, you can get benefits until your child reaches age 16. Your child's benefit will continue until he or she reaches age 18, or 19 if he or she is still in school full time. Your monthly payments stop with the child’s 16th birthday, unless your child is disabled and stays in your care.

- If you are a parent who receives most of your support from your adult child, and your child dies, Social Security also pays monthly benefits to you under the following conditions:
- You must be at least 62 years old and must not have remarried since the worker (your child)'s death
- You cannot be entitled to your own, higher Social Security benefit; and
- You must be able to show that you received one-half of your financial support from the worker at the time of their death. You must submit this proof of support to Social Security within two years of the worker's death.
We are there for those who need it most…
The Supplemental Security Income (SSI) program helps children with qualifying disabilities by providing critical financial assistance. Children and youth with specific medical conditions—whose families meet certain income and resource limits—can receive SSI from birth until age 18.
If you think your child or someone you know could be eligible for SSI, visit our webpage SSI Eligibility for Children to learn more and apply.
Assisting Youths with Disabilities Transition to Adulthood
The transition to adulthood is one of the most important periods in life’s journey. For foster children living with a disability, it can be even more challenging. Turning 18 triggers an important change in SSI benefits: Social Security must make a new determination on their SSI eligibility using the adult disability standards. About one-in-three such beneficiaries lose their SSI benefits.
For more information, please visit our spotlight page.
Apply For A Child (Under Age 18) | Disability Benefits
SSI provides monthly cash payments to help meet the basic needs of children who have a physical or mental disability or who are blind. If you care for a child or teenager with a disability, and have limited income and savings or other resources, your child may be eligible for SSI.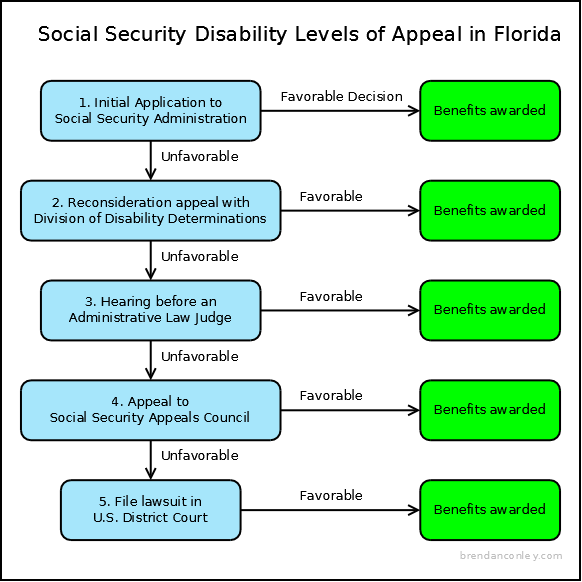
SSI Eligibility for Children
Children under age 18 can get SSI if they meet Social Security's definition of disability for children and there are limited income and resources in the household. Social Security defines a disability as:
- The child must have a physical or mental condition(s) that very seriously limits his or her activities; and
- The condition(s) must have lasted, or be expected to last, at least 1 year or result in death.
How to Apply
Tell us you want to apply for SSI for a child
Get Started
Other Ways to Apply
Apply By Phone
Call us to make an appointment to file an application at 1-800-772-1213. If you are deaf or hard of hearing, you can call us at TTY 1-800-325-0778.
Begin the Application Online
Applying for SSI requires 2 steps. You will need to complete the online Child Disability Report AND, with the help of a Social Security representative, complete an Application for SSI.
You will need to complete the online Child Disability Report AND, with the help of a Social Security representative, complete an Application for SSI.
Step 1
TIP: Before completing the Child Disability Report, use our Child Disability Starter Kit to get answers to commonly asked questions about applying for SSI. The kit also includes a worksheet that will help you gather the information you need.
Fill out the online Child Disability Report
The report usually takes about an hour to complete and collects information about the child's disabling condition and how it affects their ability to function.
We will ask you to sign a form that gives the child's doctor(s) permission to give us information about their disability. We need this information so that we can make a decision on the child's claim. In some cases, if the child is over age 12, he or she must sign his or her own medical release.
Start the Child Disability Report
If you previously started a Child Disability Report for this child but did not finish it, you can use your re-entry number to return to your online Child Disability Report.
Step 2
After you submit a report, we will call you within 3-5 business days. Together, we will:
- Review the completed Child Disability Report.
- Discuss whether the income and resources of the household are within the allowed limits.
- Start the SSI application process.
Related Questions
How can I get ready for the disability interview?
- Review the disability starter kit. It includes a checklist and a worksheet to help you gather the information you need. Have this information with you at the time of the interview.
- You can fill out a Child Disability Report.
- For more information visit Benefits for People with Disabilities or call toll-free 1-800-772-1213 (for the deaf or hard of hearing, call TTY 1-800-325-0778).

How will I know what Social Security has decided?
We will send you a letter. It can take 3 to 5 months for us to make a decision on a child’s SSI disability claim. We may also contact you by phone to ask additional questions. Let us know if your address or telephone number changes so that we can get in touch with you.
What if I am more comfortable speaking in a language other than English?
We provide free interpreter services to help you conduct your Social Security business, including helping you complete the SSI application and answering your questions. NOTE: the Child Disability Report is only available in English.
Call our toll-free number, 1-800-772-1213. If you need service in Spanish, press 7 and wait for a Spanish-speaking representative to help you. For all other languages, stay on the line and remain silent during our English voice automation prompts until a representative answers. The representative will contact an interpreter to help with your call. You may access the information on this page in Spanish.
The representative will contact an interpreter to help with your call. You may access the information on this page in Spanish.
Localized scleroderma | #05/08 | Attending Doctor is a professional medical publication for physicians. Science articles.
Over the past decade, ideas about systemic connective tissue diseases have significantly expanded, among which scleroderma occupies the second place in frequency. The disease is characterized by a systemic progressive lesion of the connective tissue with a predominance of fibrosclerotic and vascular changes in the form of obliterating endarteritis with widespread vasospastic disorders [7, 12].
Despite the lack of official statistics, it can be argued that patients with such an autoimmune disease as localized scleroderma are becoming more and more aggressive [15]. Perhaps this is due to non-compliance with the norms of medical examination and terms of treatment [11].
Discussions about the relationship between systemic (SSD) and limited (OSD) scleroderma continue. According to some authors, OSD and SJS are varieties of the same pathological process, which is confirmed by the presence of visceropathy in OSD, the unidirectionality of metabolic changes, the common pathohistological changes in the skin in both forms of the disease, as well as cases of transformation of a localized process into progressive systemic sclerosis [3, 12, 14 , eighteen]. Other researchers refer only SJS to the group of "diffuse connective tissue diseases", believing that OSD and SJS are two diseases that differ sharply in the clinical picture, course and prognosis. However, it is not always possible to draw a clear line between focal and systemic processes. Clinical observations have shown that skin lesions as one of the first signs of diffuse scleroderma are observed in 61% of cases, and descriptions of the transformation of a limited process, in particular, lichen sclerosus, into systemic scleroderma suggest the unity of these two forms. As evidenced by the results of a survey of patients with limited scleroderma, the unfavorable course of the disease with the transition to a systemic process is mainly facilitated by 4 factors:
As evidenced by the results of a survey of patients with limited scleroderma, the unfavorable course of the disease with the transition to a systemic process is mainly facilitated by 4 factors:
-
debut of the disease before the age of 20 or after 50 years;
-
multiple plaque or linear forms of the disease;
-
localization of lesions involving the skin of the face or areas above the joints of the extremities;
-
the severity of deficiency of the cellular link of immunity, dysimmunoglobulinemia, an increase in coarse circulating immune complexes and antilymphocyte antibodies [2, 3, 12, 16].
OSD, as well as SJS, are more often ill in females, for example, girls are ill more than 3 times more often than boys, and women aged 40–55 years make up 75% of patients with scleroderma [21]. The disease can occur at any age, even in newborns, usually starting without any subjective sensations and disturbance of the general condition. In connection with the tendency of the growing organism to the spread of pathology, to pronounced vascular reactions in children, this disease often has a tendency to extensive damage, although in the early stages it can manifest itself as single foci.
In connection with the tendency of the growing organism to the spread of pathology, to pronounced vascular reactions in children, this disease often has a tendency to extensive damage, although in the early stages it can manifest itself as single foci.
The pathogenesis of scleroderma is associated mainly with the hypotheses of metabolic, vascular and immune disorders. Disorders of the autonomic nervous system and neuroendocrine disorders also influence the occurrence of OSD. It is customary to consider limited scleroderma as a kind of autoimmune disease, which is based on autoimmune and inflammatory reactions to various antigens. V. A. Vladimirtsev et al. (1982) believe that an increased level of collagen proteins, being a source of active antigenic stimulation, creates a background against which, with a genetic predisposition, autoimmune reactions are realized. The emerging vicious circle of mutual influence of lymphoid and collagen-synthesizing cells leads to the progression of the fibrous process [6]. Established violations of humoral and cellular immunity in patients with scleroderma were more often recorded in women. Cellular immunity in women, in contrast to its humoral link, is less active than in men. A decrease in cellular immunity, especially its suppressor link, with an increase in the activity of humoral immunity leads to the fact that women develop an autoimmune process much more often than men. There is a link between scleroderma and pregnancy and menopause [21]. In recent years, studies have appeared on the participation of estrogen and progesterone, as well as some other hormones in the reactions of the synthesis of collagen and other components of the connective tissue. Of particular pathogenetic importance in scleroderma is microcirculation changes, which are most pronounced during menopause. They are based mainly on lesions of the walls of small arteries, arterioles and capillaries, proliferation and destruction of the endothelium, and intimal hyperplasia [3, 5, 12, 16, 20].
Established violations of humoral and cellular immunity in patients with scleroderma were more often recorded in women. Cellular immunity in women, in contrast to its humoral link, is less active than in men. A decrease in cellular immunity, especially its suppressor link, with an increase in the activity of humoral immunity leads to the fact that women develop an autoimmune process much more often than men. There is a link between scleroderma and pregnancy and menopause [21]. In recent years, studies have appeared on the participation of estrogen and progesterone, as well as some other hormones in the reactions of the synthesis of collagen and other components of the connective tissue. Of particular pathogenetic importance in scleroderma is microcirculation changes, which are most pronounced during menopause. They are based mainly on lesions of the walls of small arteries, arterioles and capillaries, proliferation and destruction of the endothelium, and intimal hyperplasia [3, 5, 12, 16, 20]. The question of the role of heredity in the development of OSD is still being discussed. According to Furst A. (2004), Oklahoma Native Americans are 8 times more likely to suffer from scleroderma than other residents of the United States. Black people are also more susceptible to this disease, they are more likely to get sick in childhood and have a more common process compared to whites. However, studies conducted by the same author found that only 6% of twins simultaneously suffer from scleroderma, and this is not a high enough incidence rate among twins to assert a purely genetic etiology of the disease.
The question of the role of heredity in the development of OSD is still being discussed. According to Furst A. (2004), Oklahoma Native Americans are 8 times more likely to suffer from scleroderma than other residents of the United States. Black people are also more susceptible to this disease, they are more likely to get sick in childhood and have a more common process compared to whites. However, studies conducted by the same author found that only 6% of twins simultaneously suffer from scleroderma, and this is not a high enough incidence rate among twins to assert a purely genetic etiology of the disease.
The contradictions in the literature data are probably due to the fact that the nature and severity of immunological, endocrine and metabolic changes largely depend on the course of the disease as a whole and on the degree of damage individually [8].
Until now, many researchers continue to support the infectious theory of the occurrence of scleroderma. The development of scleroderma may be associated with the transfer of diseases such as influenza, tonsillitis, scarlet fever, pneumonia. Some authors consider widespread scleroderma as a late manifestation of borreliosis (synonym: ixodic tick-borne borreliosis, Lyme disease), which is confirmed by the determination in some patients (especially plaque and scleroatrophic forms) of a high titer of immunoglobulin antibodies to Burgdorfer's borrelias and amazingly rapid improvement after treatment of the disease with penicillin. S. Bucher, based on the results of immunological studies and spirochete-like structures found in frozen biopsy specimens, considered this to be confirmation of the spirochete theory of the occurrence of scleroderma [9, eighteen]. Conducted observations have established various skin manifestations of Lyme borreliosis: plaque form of scleroderma (98%), Pasini-Pierini atrophoderma (80%), anetodermia and chronic atrophic acrodermatitis (100%) and rarely lichen sclerosus [4, 9, 16, 18] .
The development of scleroderma may be associated with the transfer of diseases such as influenza, tonsillitis, scarlet fever, pneumonia. Some authors consider widespread scleroderma as a late manifestation of borreliosis (synonym: ixodic tick-borne borreliosis, Lyme disease), which is confirmed by the determination in some patients (especially plaque and scleroatrophic forms) of a high titer of immunoglobulin antibodies to Burgdorfer's borrelias and amazingly rapid improvement after treatment of the disease with penicillin. S. Bucher, based on the results of immunological studies and spirochete-like structures found in frozen biopsy specimens, considered this to be confirmation of the spirochete theory of the occurrence of scleroderma [9, eighteen]. Conducted observations have established various skin manifestations of Lyme borreliosis: plaque form of scleroderma (98%), Pasini-Pierini atrophoderma (80%), anetodermia and chronic atrophic acrodermatitis (100%) and rarely lichen sclerosus [4, 9, 16, 18] .
In contrast to the above data, many researchers tend to regard cases of limited scleroderma with a high titer of antibodies to borrelia and the detection of spirochetes as borreliosis occurring under the guise of limited scleroderma, and skin sclerosis as pseudosclerotic changes, but in no case as manifestations of true scleroderma. According to N. S. Potekaev et al. (2006), the pathogenetic relationship of OSD with Lyme disease, as well as Pasini-Perini atrophoderma, Perry-Romberg syndrome, is only assumed [17]. To confirm the presence of Lyme disease in a patient with scleroderma foci, it is advisable to determine specific antibodies in the sera of patients using the methods of indirect immunofluorescence reaction (IRIF), polymerase chain reaction (PCR), as well as the detection of borreliae in skin biopsies from lesions using the silvering method [9, eighteen].
Despite the variety of theories of the occurrence of OSD, none of them reveals the initial cause and interaction of factors in the pathogenesis of the scleroderma process. The most interesting are the studies of some indicators of calcium metabolism, conducted by Bolotnaya L.A. et al. (2004). Based on the results obtained, the authors concluded that the changes in calcium and magnesium, identified at all stages of OSD, are of pathogenetic significance. The degree of these disorders is directly dependent on the activity, form and duration of dermatosis. A defect in the functions of cell membranes can cause the accumulation of calcium in various cells of patients with OSD and enhance the synthetic activity of fibroblasts, narrowing of the vessels of the microvasculature, and stimulation of lymphocytes. Hypomagnesemia, detected in these patients, contributes to the destabilization of cell membranes and may be one of the causes of calcium accumulation in erythrocytes, as well as cause dysfunction of a number of enzymes [4].
The most interesting are the studies of some indicators of calcium metabolism, conducted by Bolotnaya L.A. et al. (2004). Based on the results obtained, the authors concluded that the changes in calcium and magnesium, identified at all stages of OSD, are of pathogenetic significance. The degree of these disorders is directly dependent on the activity, form and duration of dermatosis. A defect in the functions of cell membranes can cause the accumulation of calcium in various cells of patients with OSD and enhance the synthetic activity of fibroblasts, narrowing of the vessels of the microvasculature, and stimulation of lymphocytes. Hypomagnesemia, detected in these patients, contributes to the destabilization of cell membranes and may be one of the causes of calcium accumulation in erythrocytes, as well as cause dysfunction of a number of enzymes [4].
There is no single generally accepted classification of OSD; in our opinion, the classification of S. I. Dovzhansky (1979) is more acceptable, in which all clinical forms of OSD are most fully represented:
-
Plaque (discoid):
a) indurative-atrophic;
b) surface "lilac";
c) nodular, deep;
d) bullous;
e) generalized.
-
Linear:
a) by the type of "saber strike";
b) ribbon-like, strip-like;
c) zosteriform.
-
3. White spot disease.
-
4. Pasini-Pierini idiopathic atrophoderma.
Excessive fibrosis and impaired microcirculation form the clinical picture of the disease, the features of its manifestations. Among all clinical varieties of OSD, the most common form is plaque. Plaque scleroderma is characterized by the formation of a small number of rounded lesions. Foci in their development go through three stages: spots, plaques and atrophy. The disease begins imperceptibly with the appearance of one or more lilac-pink rounded spots of various sizes. Gradually, the center of the spots turns pale and begins to thicken, the lesion eventually turns into a very dense plaque of a characteristic yellowish-white color (“ivory”) with a smooth, shiny surface. On the periphery of the plaques, a lilac halo persists for some time, due to which they grow and which can be used to judge the activity of the process. Hair on the plaques falls out, the skin pattern is smoothed out, sweat and sebum secretion stops; the skin on the affected area cannot be gathered into a fold. In this state, the foci can remain indefinitely, and then gradually undergo atrophy [2, 10, 19].
Hair on the plaques falls out, the skin pattern is smoothed out, sweat and sebum secretion stops; the skin on the affected area cannot be gathered into a fold. In this state, the foci can remain indefinitely, and then gradually undergo atrophy [2, 10, 19].
A rarer type of localized scleroderma is the band-like (linear) type, usually seen in children. The difference from plaque scleroderma is only in the outlines of the foci - they look like stripes and are usually located on the limbs and along the sagittal line on the forehead (reminiscent of a scar from a saber strike).
Another type of scleroderma is lichen scleroatrophic (teardrop scleroderma, white spot disease, white Tsumbush lichen). It is suggested that it may be an atrophic form of lichen planus, or kraurosis; the independence of dermatosis is not excluded. However, a combination of plaque scleroderma and scleroatrophic lichen is often observed, which indicates the unity of these clinical forms. Rashes in lichen sclerosus are represented by small scattered or grouped whitish spots, sometimes with a liquid tint, 0.5–1.5 cm in size, more often localized on the skin of the trunk and neck, as well as on any part of the skin. Often the disease develops in girls and young women in the genital area. There are common forms of scleroatrophic lichen and atypical variants; bullous and telangiectatic [2, 10, 19]. The bullous form is characterized by the formation of blisters with a dense cover and serous contents. Blisters may open, exposing erosions, or shrink into a dense serous crust. Bubbles indicate the progression of the atrophic process, and if erosion and ulcers subsequently form in their place, the process is difficult to treat. In the telangiectatic form, telangiectasias form in areas of whitish atrophy of the skin [12].
Rashes in lichen sclerosus are represented by small scattered or grouped whitish spots, sometimes with a liquid tint, 0.5–1.5 cm in size, more often localized on the skin of the trunk and neck, as well as on any part of the skin. Often the disease develops in girls and young women in the genital area. There are common forms of scleroatrophic lichen and atypical variants; bullous and telangiectatic [2, 10, 19]. The bullous form is characterized by the formation of blisters with a dense cover and serous contents. Blisters may open, exposing erosions, or shrink into a dense serous crust. Bubbles indicate the progression of the atrophic process, and if erosion and ulcers subsequently form in their place, the process is difficult to treat. In the telangiectatic form, telangiectasias form in areas of whitish atrophy of the skin [12].
Vulvar lichen sclerosus (SALV) is considered a rare disease, but the disease is not as rare in children as it follows from foreign literature data. Most children (70%) fall ill before the age of 10–11 years, that is, before the onset of puberty. SALV is considered a disease with unknown etiology and pathogenesis; some authors note the involvement of a hormonal factor in its pathogenesis. In particular, E. A. Burova (1989) indicates the leading role of dyshormonal disorders in the pituitary-adrenals-ovaries system. The clinical picture of SALV is represented by the formation of small whitish-grayish scleroatrophic foci, sometimes with a pearly tint, luster, punctate depressions, follicular keratosis, and a lilac edge. Atrophic changes are most pronounced when localized in the vulva. Girls with SALV due to low estrogen levels are characterized by later puberty, menstrual dysfunction [2, 5, 10, 19].
Most children (70%) fall ill before the age of 10–11 years, that is, before the onset of puberty. SALV is considered a disease with unknown etiology and pathogenesis; some authors note the involvement of a hormonal factor in its pathogenesis. In particular, E. A. Burova (1989) indicates the leading role of dyshormonal disorders in the pituitary-adrenals-ovaries system. The clinical picture of SALV is represented by the formation of small whitish-grayish scleroatrophic foci, sometimes with a pearly tint, luster, punctate depressions, follicular keratosis, and a lilac edge. Atrophic changes are most pronounced when localized in the vulva. Girls with SALV due to low estrogen levels are characterized by later puberty, menstrual dysfunction [2, 5, 10, 19].
Atrophoderma Pasini-Pierini is characterized by a few spots that are located mainly on the back and are usually large (up to 10 cm or more) and often irregular in shape. The disease is, as it were, a transitional form between plaque scleroderma and skin atrophy. This variety is usually seen in young women. Eruptions - in the form of bluish-purple spots with a smooth, slightly sunken center, but without the phenomenon of falling through the finger or hernial protrusion. Sometimes a lilac ring is visible around the spot. A characteristic feature of this form of OSD is the prolonged absence of compaction at the onset of the disease. In some cases, pigmentation is clearly expressed. Simultaneously with the clinical manifestations of Pasini-Pierini atrophoderma, typical manifestations of OSD can be observed [2, 10, 19]. Although it is believed that atrophoderma is an independent disease, it seems to be more correct to consider it as a clinical type of OSD, especially since in some cases atrophy and hyperpigmentation precede the development of sclerosis, which still appears on atrophoderma plaques only after a few years. The difference between idiopathic atrophoderma and plaque scleroderma is that with atrophoderma, the skin of the trunk, and not the face and limbs, is mainly affected, and the process itself develops for a long time (for several years), the lesions are plaques almost without compaction, bluish-brown in color without a purple ring around the periphery.
This variety is usually seen in young women. Eruptions - in the form of bluish-purple spots with a smooth, slightly sunken center, but without the phenomenon of falling through the finger or hernial protrusion. Sometimes a lilac ring is visible around the spot. A characteristic feature of this form of OSD is the prolonged absence of compaction at the onset of the disease. In some cases, pigmentation is clearly expressed. Simultaneously with the clinical manifestations of Pasini-Pierini atrophoderma, typical manifestations of OSD can be observed [2, 10, 19]. Although it is believed that atrophoderma is an independent disease, it seems to be more correct to consider it as a clinical type of OSD, especially since in some cases atrophy and hyperpigmentation precede the development of sclerosis, which still appears on atrophoderma plaques only after a few years. The difference between idiopathic atrophoderma and plaque scleroderma is that with atrophoderma, the skin of the trunk, and not the face and limbs, is mainly affected, and the process itself develops for a long time (for several years), the lesions are plaques almost without compaction, bluish-brown in color without a purple ring around the periphery. Complete regression of atrophoderma is not observed, while the focus of plaque scleroderma may disappear completely (with timely treatment), or after it there remains slight atrophy or persistent pigmentation [2, 10, 19].
Complete regression of atrophoderma is not observed, while the focus of plaque scleroderma may disappear completely (with timely treatment), or after it there remains slight atrophy or persistent pigmentation [2, 10, 19].
One of the rare manifestations of scleroderma is Parry-Romberg face hemiatrophy, a disease characterized by progressive atrophy of only one half of the face, manifested by dystrophic changes in the skin and subcutaneous tissue, and to a lesser extent, muscles and the facial skeleton. The general condition of patients, as a rule, remains satisfactory, the main complaint is a cosmetic defect in the face. According to the literature, women predominate among patients. In most cases, the disease develops between the ages of 3 and 17 years. Left-sided and right-sided localization of the process is equally often noted. As a rule, the disease has a long, chronic course. The active stage lasts mainly up to 20 years, in some observations - up to 40 years. The first signs of the disease are local changes in the skin of the face, which soon acquires a yellowish or bluish tint. Gradually develops thickening of the skin in the foci. In the future, in places of compaction, the skin atrophies; over time, atrophic changes progress with the involvement of subcutaneous adipose tissue and facial muscles in the process. The most pronounced and frequent signs of skin lesions are its sharp thinning, wrinkling, diffuse or focal hyperpigmentation. In atrophied areas of the skin, there is no hair growth. In patients, not only the skin suffers, but also the underlying soft tissues, which, as a rule, leads to gross deformity of the face in the form of a significant asymmetry of its right and left halves, most pronounced at the onset of the disease in early childhood. Bone structures are also affected if hemiatrophy occurs before the end of their growth. In some patients, atrophy of half of the tongue is observed. There are clinical observations of the development of progressive hemiatrophy of the face as a manifestation of the terminal stage in patients with an aggressive course of strip-like scleroderma on the face.
The first signs of the disease are local changes in the skin of the face, which soon acquires a yellowish or bluish tint. Gradually develops thickening of the skin in the foci. In the future, in places of compaction, the skin atrophies; over time, atrophic changes progress with the involvement of subcutaneous adipose tissue and facial muscles in the process. The most pronounced and frequent signs of skin lesions are its sharp thinning, wrinkling, diffuse or focal hyperpigmentation. In atrophied areas of the skin, there is no hair growth. In patients, not only the skin suffers, but also the underlying soft tissues, which, as a rule, leads to gross deformity of the face in the form of a significant asymmetry of its right and left halves, most pronounced at the onset of the disease in early childhood. Bone structures are also affected if hemiatrophy occurs before the end of their growth. In some patients, atrophy of half of the tongue is observed. There are clinical observations of the development of progressive hemiatrophy of the face as a manifestation of the terminal stage in patients with an aggressive course of strip-like scleroderma on the face. The literature provides data on the results of examination of patients with scleroderma, 16.7% of which subsequently developed facial hemiatrophy. Such cases suggest that Romberg's hemiatrophy is an unfavorable variant of the course of OSD [1].
The literature provides data on the results of examination of patients with scleroderma, 16.7% of which subsequently developed facial hemiatrophy. Such cases suggest that Romberg's hemiatrophy is an unfavorable variant of the course of OSD [1].
Diagnosis of limited scleroderma can present certain difficulties, especially in the initial stage of the disease. The differential diagnosis of OSD is carried out with vitiligo, kraurosis, undifferentiated form of leprosy, Shulman's syndrome.
At the beginning of the development of plaque scleroderma, when the seal is not yet expressed and there is only a discolored spot, the process may resemble vitiligo or a depigmented spot in undifferentiated leprosy. With vitiligo, the spots have a clearer border, which is clearly visible in the presence of a hyperpigmented zone. The surface of the spots is smooth, without signs of atrophy and peeling. Vitiligo spots persist for quite a long time without compaction.
In undifferentiated leprosy, skin changes are characterized by patchy rashes. The latter may be erythematous, of various shades (from pink to cyanotic) and hypopigmented. Pain, tactile and temperature sensitivity is reduced in the area of spots.
It is more difficult to differentiate linear scleroderma from a linearly located keloid-like nevus. A distinctive feature may be the detection of a keloid-like nevus in the first months of life and its long existence without pronounced changes over many years.
Kraurosis of the vulva may to some extent resemble scleroderma, in particular lichen sclerosus, since in this disease the surface of the affected areas is dry, shiny, and dense. However, vulvar kraurosis has intense itching and telangiectasias. In the future, atrophy of the labia minora and labia majora, leukoplakia and, often, cancer develop. Kraurosis of the penis manifests itself in the form of chronic atrophy and wrinkling of the glans penis and the inner layer of the foreskin, while sclerotic changes in the foreskin and glans penis cause phimosis and narrowing of the urethra. In contrast to vulvar kraurosis, penile lesions do not itch and the disease is not complicated by cancer.
In contrast to vulvar kraurosis, penile lesions do not itch and the disease is not complicated by cancer.
Shulman syndrome (syn.: eosinophilic fasciitis, diffuse fasciitis with hypergammaglobulinemia and eosinophilia). This disease is understood as diffuse scleroderma-like thickening of the skin, thickening of the muscular fascia, its infiltration with eosinophils, lymphocytes and plasma cells. Foci are localized more often on the limbs and lead to flexion contractures. The differential signs characteristic of Shulman's syndrome are the absence of a purple corolla around the focus of induration and skin atrophy, as well as the presence of pain and eosinophilia in the peripheral blood [2, 10, 19].
When localizing the focus of scleroderma on the face, it is necessary to remember such a rare type of tumor as the scleroderma-like form of basalioma, in which the nodule pathognomonic for basalioma slowly increases in size, transforms into a dense, ivory-colored plaque slightly rising above the skin surface with a waxy sheen, in the central part showing telangiectasia. The boundaries of the focus are sharp, the outlines are rounded or irregular, the sizes are from 1 to 3 cm or more. This form is difficult to diagnose if the nodules present on the periphery of the focus, pathognomonic for basalioma, are ignored.
The boundaries of the focus are sharp, the outlines are rounded or irregular, the sizes are from 1 to 3 cm or more. This form is difficult to diagnose if the nodules present on the periphery of the focus, pathognomonic for basalioma, are ignored.
Treatment. OSD therapy should be multi-course and multicomponent. With an active process, the number of courses should be at least 6, with an interval of 1-2 months; if the process has stabilized, the interval between courses of treatment increases to 4 months; with residual clinical manifestations and for the purpose of prevention, therapy is carried out 2-3 times a year with drugs that improve microcirculation.
With an active process, the following groups of drugs should be included in the treatment of OSD:
-
Antibiotics of the penicillin series - the recommended course is 15–20 million units. In case of intolerance to penicillin, it can be replaced with fusidic acid, oxacillin, ampicillin, amoxicillin, which must be prescribed under the guise of antihistamines.
 The first 3 courses of treatment must include antibiotics.
The first 3 courses of treatment must include antibiotics. -
An important drug in the treatment of the disease is Lidaza, which contains hyaluronidase, which breaks down hyaluronic acid, which is the cement of the connective tissue. In addition, Lidaza increases the permeability of tissues and facilitates the movement of fluid in interstitial spaces. Lidase can be replaced with aloe or placenta extract, Collalizin, Actinogyal, Longidase.
-
Vascular funds. First of all, these are nicotinic acid preparations that have a vasodilating effect, improve carbohydrate metabolism, and have hypocholesterolemic activity (reduce triglycerides and lipoproteinemides). Xanthinol nicotinate (Teonicol, Complamin) - is a combination of theophylline and nicotinic acid, dilates peripheral vessels, improves cerebral circulation, and reduces platelet aggregation. It is important to know that xanthinol nicotinate is not recommended for use in the first trimester of pregnancy, with a stomach ulcer; it also cannot be combined with antihypertensive drugs.
 Also drugs of choice are Trental, Mildronate. When choosing a therapeutic drug, one should not forget about herbal remedies that improve microcirculation.
Also drugs of choice are Trental, Mildronate. When choosing a therapeutic drug, one should not forget about herbal remedies that improve microcirculation.
A certain therapeutic effect was obtained from injections of Madecassol (a preparation from Asian cintella) for 2-4 weeks of use, Madecassol has a less pronounced antifibrotic, but positive vascular effect. Experimental studies have established the suppression of the biosynthesis of collagen and the components of the basic substance of the connective tissue, the slowing down of fibrosis under the influence of the drug. Madecassol is most effective in patients with widespread OSD, in which the simultaneous administration of the drug in tablets and in the form of an ointment is indicated. Also from phytopreparations it is recommended Piascledin, Aescusan, Berberine 1 tab. 3 times a day for 1-2 months. -
Antagonists of calcium ions. The drugs of this group have a specific ability to inhibit the penetration of calcium ions into myofibrils and thereby reduce the activity of myofibrillar ATPase.
 They cause relaxation of muscle fibers and reduce resistance in the coronary and peripheral vessels. Calcium ion antagonists improve coronary blood flow and oxygen supply to the heart, dilate peripheral vessels and cause some decrease in systemic arterial pressure. The drugs in this group include nifedipine (Corinfar, Fenigidin, Calcigard retard), verapamil. Bolotnaya L.A. (2004) proved the clinical efficacy of calcium antagonists in the treatment of OSD using the example of cinnarizine (Stugeron). “Natural physiological calcium blockers” also include preparations containing magnesium (Magne B6) [3].
They cause relaxation of muscle fibers and reduce resistance in the coronary and peripheral vessels. Calcium ion antagonists improve coronary blood flow and oxygen supply to the heart, dilate peripheral vessels and cause some decrease in systemic arterial pressure. The drugs in this group include nifedipine (Corinfar, Fenigidin, Calcigard retard), verapamil. Bolotnaya L.A. (2004) proved the clinical efficacy of calcium antagonists in the treatment of OSD using the example of cinnarizine (Stugeron). “Natural physiological calcium blockers” also include preparations containing magnesium (Magne B6) [3].
In case of scleroatrophic lichen, it is recommended to add Retinol palmitate 100,000 IU per day to the treatment, and locally Solcoseryl, Actovegin ointments, creams with vitamin E, F.
With single lesions, one can limit oneself to the appointment of vitamin B12 in suppositories and phonophoresis with Lidaza, Ronidase, trypsin, chemotripsin (No. 7–10). Local treatment of OSD should consist of topical applications and physiotherapy. In the topical therapy of OSD, the following ointments are usually used: Heparin, Heparoid, Troxovasin, Butadion, Theonicol. The drugs of choice for topical therapy are Dimexide, Unitiol, Ronidase, trypsin, chymotrypsin, Lidaza, which can be used as applications or injected into lesions using electro- and phonophoresis. Ronidase is applied externally by applying its powder (0.5–1.0 g) to a napkin moistened with saline. Apply a napkin to the lesion, fixing with a bandage for half a day. The course of applications continues for 2-3 weeks. It is recommended to prescribe Kuprenil and Hydrocortisone ultraphonophoresis to the lesions. In OSD, magnetotherapy, vacuum decompression, and low-intensity laser therapy are also used. At the end of the course of therapy, you can attach a massage of the lesions. With a decline in the activity of the process - hydrogen sulfide and radon baths [10, 19, twenty].
7–10). Local treatment of OSD should consist of topical applications and physiotherapy. In the topical therapy of OSD, the following ointments are usually used: Heparin, Heparoid, Troxovasin, Butadion, Theonicol. The drugs of choice for topical therapy are Dimexide, Unitiol, Ronidase, trypsin, chymotrypsin, Lidaza, which can be used as applications or injected into lesions using electro- and phonophoresis. Ronidase is applied externally by applying its powder (0.5–1.0 g) to a napkin moistened with saline. Apply a napkin to the lesion, fixing with a bandage for half a day. The course of applications continues for 2-3 weeks. It is recommended to prescribe Kuprenil and Hydrocortisone ultraphonophoresis to the lesions. In OSD, magnetotherapy, vacuum decompression, and low-intensity laser therapy are also used. At the end of the course of therapy, you can attach a massage of the lesions. With a decline in the activity of the process - hydrogen sulfide and radon baths [10, 19, twenty].
Current trends towards a reduction in the volume of drug therapy contribute to the introduction of drugs that combine several therapeutic effects. Systemic polyenzymes, which are a stable mixture of enzymes of plant and animal origin, rutin, have such a multifactorial effect on the body. The rationale for the use of Wobenzym in the treatment of OSD (in tablets and in the form of an ointment) was its effect on collagen metabolism, the ability to suppress the formation and breakdown of pathological immune complexes, increase the cytotoxic activity of macrophages, improve microcirculation due to the effect on platelets and blood rheology, maintain normal functioning of endogenous enzymes. Starting dose 5 tab. 3 times a day against the background of the main therapy, then according to indications: 3-4 tab. 3 times a day.
Systemic polyenzymes, which are a stable mixture of enzymes of plant and animal origin, rutin, have such a multifactorial effect on the body. The rationale for the use of Wobenzym in the treatment of OSD (in tablets and in the form of an ointment) was its effect on collagen metabolism, the ability to suppress the formation and breakdown of pathological immune complexes, increase the cytotoxic activity of macrophages, improve microcirculation due to the effect on platelets and blood rheology, maintain normal functioning of endogenous enzymes. Starting dose 5 tab. 3 times a day against the background of the main therapy, then according to indications: 3-4 tab. 3 times a day.
In recent years, hyperbaric oxygen therapy (HBO) has been widely used in the treatment of various pathologies, which contributes to a more intensive enrichment of tissues with oxygen, which at elevated pressure can also have an antimicrobial effect, especially against anaerobic microorganisms. HBO increases the metabolic activity of mitochondria and their ability to regenerate, normalizes lipid oxidation, increases the level of oxygen utilization by tissues due to the activation of aerobic processes in the lesions, improves microcirculation. The expediency of using HBO in scleroderma is indicated by a number of authors [3, 13].
HBO increases the metabolic activity of mitochondria and their ability to regenerate, normalizes lipid oxidation, increases the level of oxygen utilization by tissues due to the activation of aerobic processes in the lesions, improves microcirculation. The expediency of using HBO in scleroderma is indicated by a number of authors [3, 13].
Many authors recommend plasma-substituting dextran preparations (Dextran, Rheomacrodex) [3, 10, 16]. However, in our opinion, such therapy should be used for common, rapidly progressive forms of OSD.
We have not noted a significant effect of treatment with hormones, anabolics and quinoline drugs.
In conclusion, I would like to emphasize that the treatment of patients with OSD should be approached individually, depending on the stage of the process, prevalence, and the presence of concomitant diseases. It is necessary to explain to the patient the feasibility of long-term therapy, careful examination and preventive treatment.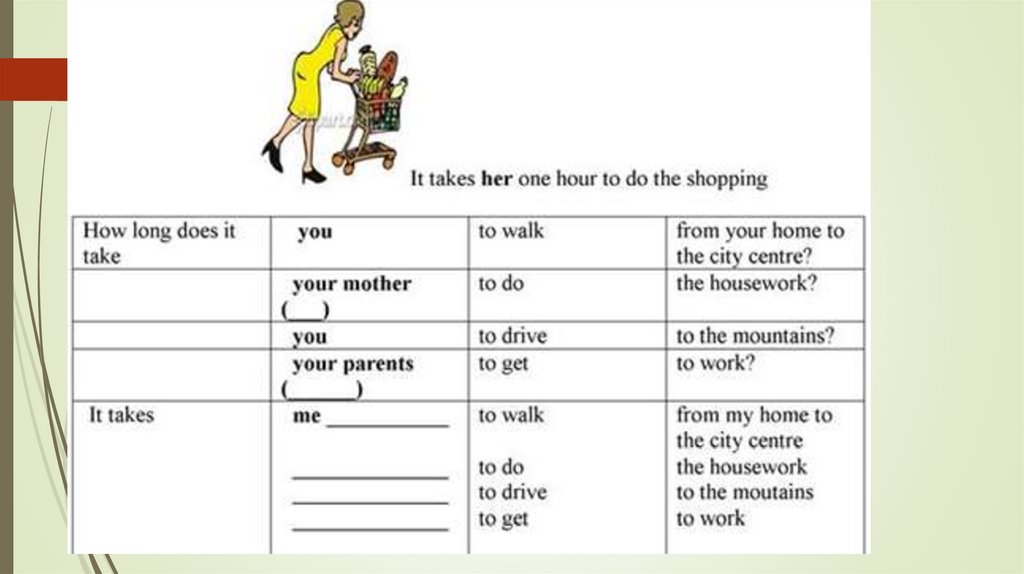
Literature
-
Belousova T. A., Kolmogorova I. V., Mokina E. V. Romberg face hemiatrophy: a modern view of the problem // Russian Journal of Skin and Venereal Diseases. 1999, No. 5, pp. 20–23.
-
Berenbein B. A., Studnitsin A. A. Differential diagnosis of skin diseases. Moscow: Medicine, 1989. 672 p.
-
Bolotnaya L. A., Serbina I. M. Modern pathogenetic therapy of scleroderma // International Medical Journal. 1999, No. 3, pp. 56–58.
-
Bolotnaya L. A., Shakhova F. B., Serbina I. M. New in the pathogenesis and therapy of limited scleroderma // Bulletin of dermatology and venereology. 2004, No. 2, pp. 31–34.
-
Burova E. A. Vulvar lichen sclerosus in children, clinical features, pathogenesis, treatment // Dis. ... cand. honey. Sciences. M., 1989. 110 p.
-
Vladimirtsev V.A., Avdeeva Zh.I., Guseva N.G. et al. Study of cellular immune response to type 1 collagen in patients with systemic scleroderma // Vopr.
 rheumatism 1982, No. 1, pp. 33–38.
rheumatism 1982, No. 1, pp. 33–38. -
Guseva N. G. Systemic scleroderma: clinic, diagnosis, treatment // Ros. magazine leather and veins. diseases. 2002, No. 4, pp. 5–15.
-
Dovzhansky S. I. Clinical and immunological parallels in limited and systemic scleroderma // Ros. magazine skin. and veins. Bol. 2002, No. 4, pp. 26–29.
-
Domaseva T. V., Babkin A. V. The spectrum of skin atrophy in Lyme borreliosis // Eighth All-Russian Congress of Dermatovenereologists. Abstracts of scientific works, part 1. Dermatology. Moscow, 2001, p. 176.
-
Ivanov O. L. Skin and venereal diseases // Handbook. Moscow: Medicine, 1997. 352 p.
-
Korobeynikova E. A., Martynova L. M., Anisimova A. V. Clinical aspects of limited scleroderma // Ros. magazine skin. and veins. Bol. 2004, No. 3, pp. 27–29.
-
Kryazheva S. S., Boldyreva M. V. Telangiectatic form of lichen sclerosus // Russian Journal of Skin and Venereal Diseases.
 1996, No. 6, pp. 27–29.
1996, No. 6, pp. 27–29. -
Kryazheva S. S., Sapronova T. I., Bulokhova L. M. Hyperbaric oxygenation in the complex therapy of plaque scleroderma // Russian Journal of Skin and Venereal Diseases. 1998, No. 4, pp. 39–41.
-
Kryazheva S.S., Sapronova T.I., Bulokhova L.M. On the problem of transformation of limited skin forms of scleroderma into systemic // Russian Journal of Skin and Venereal Diseases. 1998, No. 6, pp. 10–13.
-
Kubanova A. A., Tikhonova L. I. Dermatovenereology in Russia. Reality and prospects // Bulletin of dermatology and venereology. 2004, No. 2, pp. 4–11.
-
Ponomarev A. A., Kulikov E. P., Karavaev N. S., Fedoseev A. V. Rare skin-visceral syndromes. Ryazan, 1998. 648 p.
-
Potekaev N.S., Potekaev N.N. Lyme disease and skin lesions caused by it // Bulletin of dermatology and venereology. 2006, No. 6. S. 3–9.
-
Samsonov V. A., Olisova M. O.
 , Milonova T. I. On the relationship between Lyme disease and focal scleroderma // Bulletin of Dermatology and Venereology. 1996, No. 1. S. 8–9.
, Milonova T. I. On the relationship between Lyme disease and focal scleroderma // Bulletin of Dermatology and Venereology. 1996, No. 1. S. 8–9. -
Skripkin Yu.K. Skin and venereal diseases. A guide for doctors in 4 volumes. T. 2 / Edited by Yu. K. Skripkin. Moscow: Medicine, 1995. 544 p.
-
Smirnov A. V., Glavinskaya T. A. Modern ideas about the pathogenesis and treatment options for limited scleroderma // Nizhny Novgorod medical journal. 1997, No. 3, pp. 73–82.
-
Furst A. Scleroderma: a fascinating, troubling disease // Advanced Practice Nursing journal. 2004. 4(2).
Yu. A. Galliamova Doctor of Medical Sciences, Associate Professor
RMAPO
What you need to know about disk defragmentation in Windows, macOS and Linux
February 11, 2021LikbezDevices But it doesn't always have to be done.
Share
0Why Disk Defragmentation Is Necessary
When you move, copy, delete, and do other things with data on your hard disk drive (HDD), that data becomes fragmented. The system separates files into parts and saves them in different physical areas of the hard drive.
The system separates files into parts and saves them in different physical areas of the hard drive.
A drive that has a lot of fragmented files becomes slow. The fact is that a mechanical head is used to read a regular hard disk, which runs from one piece of data to another. The higher the fragmentation, the more operations it takes to read and the more time it takes to read. Moreover, such intensive use of the disk accelerates the wear of the drive.
Fragmentation problems are solved by the reverse process - defragmentation, during which the system moves parts of fragmented files closer to each other.
Ask any advanced user how to make a computer faster, and he will probably start talking about disk defragmentation. In the past, this advice was indeed relevant, but these days, things are a little different.
Do you need disk defragmentation in Windows
Simple answer: if you are using a more or less modern computer with an up-to-date operating system, then no, you don't need it. However, if you have an old computer with Windows XP installed on the HDD, then defragmenting it can slightly increase its performance. Let's deal with everything in order.
However, if you have an old computer with Windows XP installed on the HDD, then defragmenting it can slightly increase its performance. Let's deal with everything in order.
If you are running Windows on an SSD
SSDs, i.e. Solid State Drives, do not need to be defragmented. Such disks have no moving parts at all, so their speed does not depend on the level of fragmentation. Moreover, defragmentation can harm the SSD. This procedure repeatedly overwrites the files on the disk, which accelerates the wear of SSDs.
Modern Windows systems are smart enough not to defragment SSDs automatically. And third-party programs usually warn about the consequences.
If you are using Windows Vista, 7, 8, 10 on an HDD
Even if your system is placed on a hard drive in the old manner, you do not need to defragment it yourself. Starting with Vista, Windows does this automatically by default in the background. Usually once a week, at 1 am every Wednesday.
You can verify this and check the defragmentation settings. Open your computer, right-click on the local drive and select Properties → Tools → Optimize.
In the "Optimize disks" window, click "Change settings" and make sure that automatic defragmentation is active and runs weekly.
If you are using Windows XP on an HDD
As mentioned above, defragmentation can slightly increase device performance in this case. However, Windows XP unfortunately does not have an automatic defragmenter, which is not surprising given the age of the system.
However, the operation can still be performed manually. Open "My Computer" and right-click the system drive. Then click Properties → Tools → Run Defrag → Defrag and wait.
You can also automatically defragment your drive using third-party software. By installing a free utility like Defraggler, you can set up a schedule to run the process regularly.
The program interface is very simple and available in Russian, so it is easy to understand it. Enable weekly defragmentation in Defraggler settings and the utility will take care of your disks.
Enable weekly defragmentation in Defraggler settings and the utility will take care of your disks.
Do you need disk defragmentation in macOS
macOS is designed differently than Windows, so Mac hard drives do not need to be manually defragmented. The system optimizes drives on its own.
So, even if you use an Apple computer with an HDD, you should not worry about defragmenting its disk. And with modern Macs with SSD, this question is all the more eliminated.
But be aware that if there is less than 10% of unallocated space on your Mac hard drive, the system may have problems auto-optimizing. So make sure macOS always has free space.
Do you need disk defragmentation in Linux
The answer is the same as in the case of macOS. Linux distributions' ext4 and Btrfs filesystems are smarter than Windows' NTFS and allocate files on disk in a more advanced way. In addition, periodically the system optimizes the disk itself. Therefore, Linux does not need to be defragmented.