Cystic fibrosis and infants
Cystic fibrosis and your baby
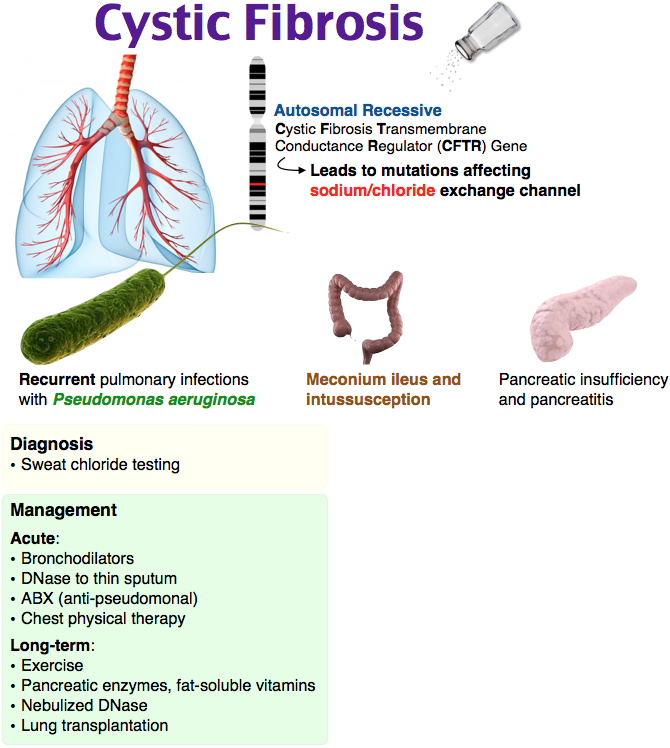
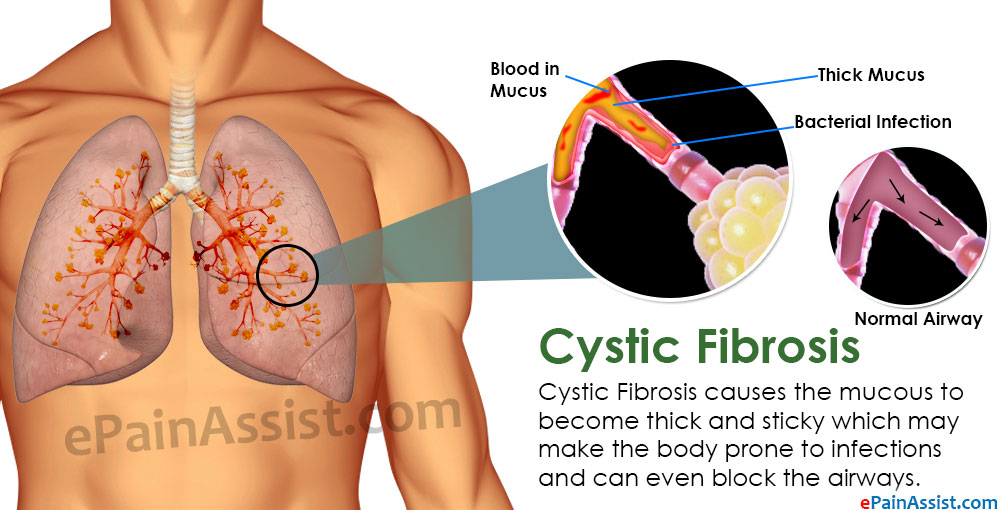
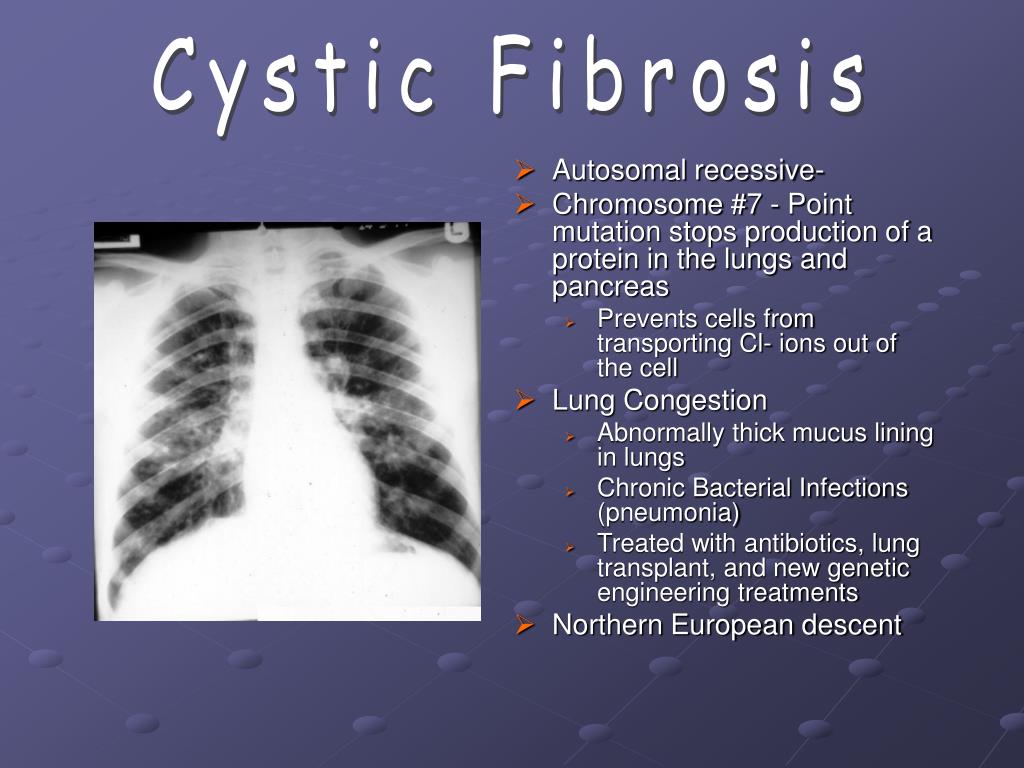
Cystic fibrosis (also called CF) is a condition that causes thick mucus to build up in the body. This causes problems with breathing and digestion.
CF is passed from parents to children through genes. A baby has to inherit a CF gene from both parents to have CF.
All babies have a newborn screening test for CF so it can be found and treated early.
Treatment can include medicines and chest therapy to help with your baby’s breathing and digestion.
Cystic fibrosis (CF) is a condition that affects breathing and digestion. It’s caused by very thick mucus that builds up in the body.
Mucus is a fluid that normally coats and protects parts of the body. It’s usually slippery and slightly thicker than water. But in CF, the mucus is thicker and sticky. It builds up in the lungs and digestive system and can cause problems with how you breathe and digest food.
CF affects about 30,000 children and adults in the United States. It is one of the most common genetic conditions in this country. CF is more common in white babies (about 1 in 3,500) than in Hispanic, Native American or Alaskan Native babies (about 1 in 10,000), in Black babies (about 1 in 15,000 black) and in Asian babies (about 1 in 30,000).
What causes CF?
CF is inherited. This means it’s passed from parent to child through genes. A gene is a part of your body’s cells that stores instructions for the way your body grows and works. Genes come in pairs—you get one of each pair from each parent.
Sometimes the instructions in genes change. This is called a gene change or a mutation. Parents can pass gene changes to their children. Sometimes a gene change can cause a gene to not work correctly. Sometimes it can cause birth defects or other health conditions. A birth defect is a health condition that is present in a baby at birth.
Your baby has to inherit a gene change for CF from both parents to have CF. If they inherit the gene change from just one parent, they have the gene change for CF, but they doesn’t have the condition.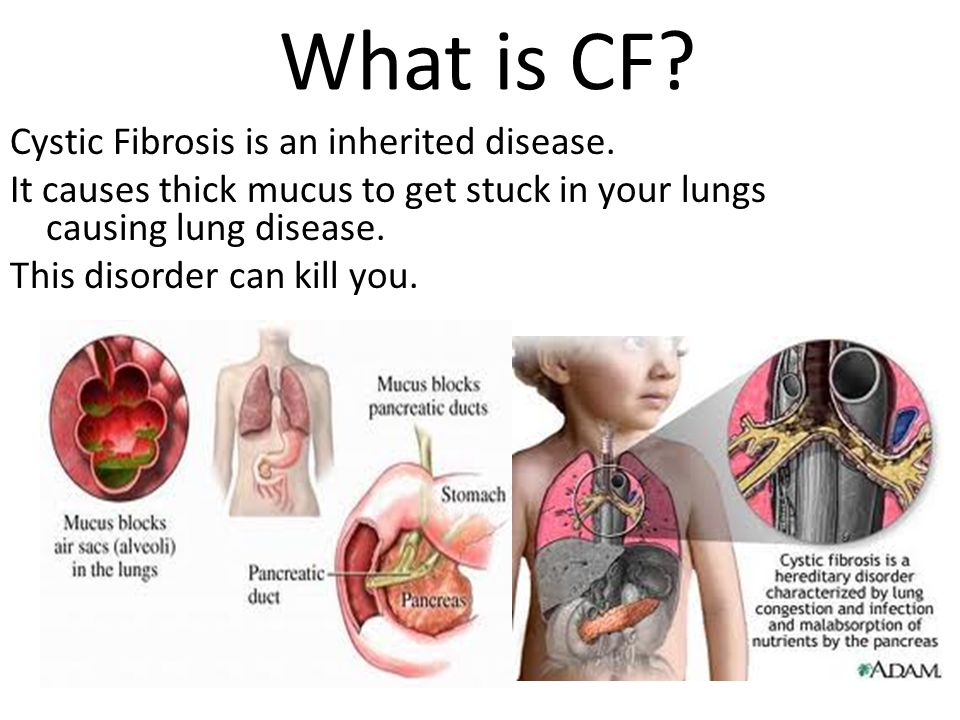 When this happens, your baby is called a CF carrier.
When this happens, your baby is called a CF carrier.
What problems does CF cause?
Babies who have CF have very thick and sticky mucus that builds up in the body. When this mucus builds up in the lungs, it blocks airways and causes breathing problems and infections. Airways are tubes that carry air in and out of the lungs. As a baby with CF gets older, lung infections can get worse. This can lead to serious, and sometimes deadly, lung damage.
When mucus builds up in the digestive system, it blocks tubes in the pancreas, an organ in the belly. This can make it hard for the body’s digestives system to break down food. When this happens, your baby may not get the nutrients they need to grow and stay healthy.
Some cases of CF are more serious than others. Babies who have CF are often sick with infections and need a lot of special medical care.
How do you know if your baby has CF?
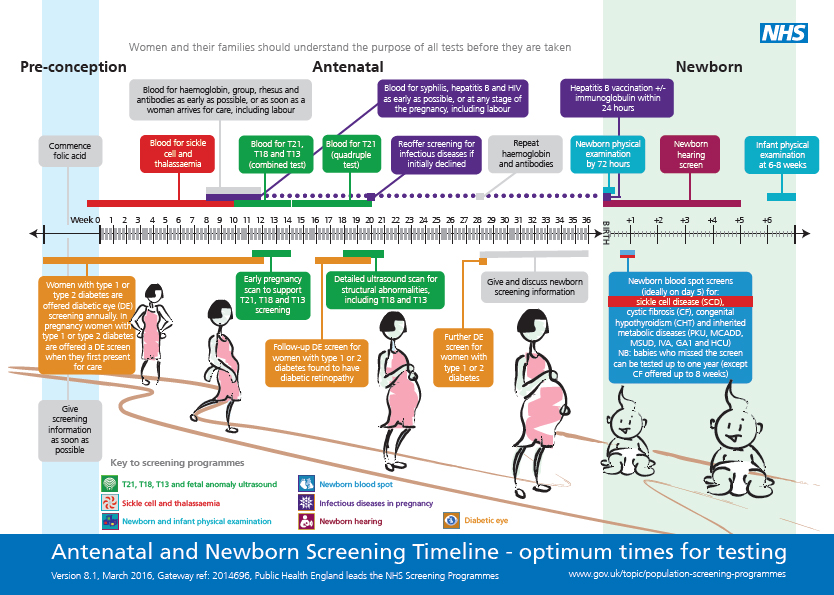
All babies have newborn screening tests for CF.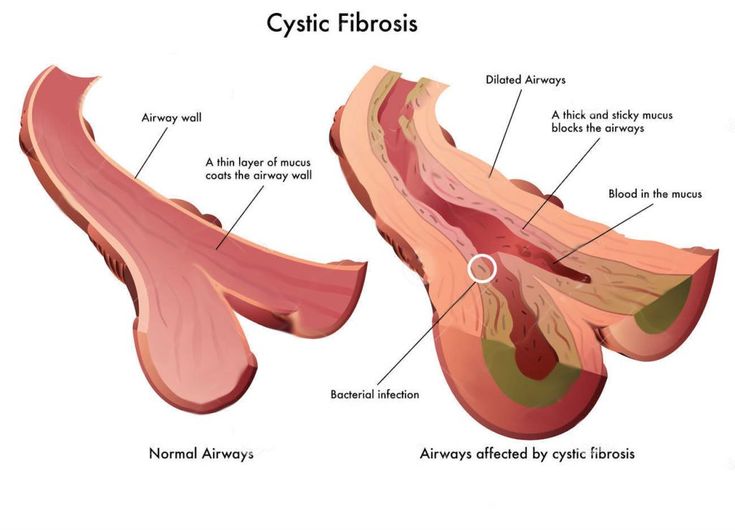 With newborn screening tests, CF can be found and treated early.
With newborn screening tests, CF can be found and treated early.
Before your baby leaves the hospital, their health care provider takes a few drops of blood from their heel to test for CF and other conditions. The blood is collected and dried on a special paper and sent to a lab for testing.
If newborn screening results aren’t normal, it simply means your baby needs more testing. Your baby’s provider can recommend another kind of test, called a diagnostic test. This test can check to see if your baby has CF or if there is some other cause for abnormal test results.
Your provider may recommend that your baby have a sweat test to see if they have CF. This is a simple, painless test that checks the amount of salt in your baby’s sweat. Babies with CF have more salt in their sweat than healthy babies. Your baby’s provider also may recommend a genetic test for your baby.
If your baby does have CF, they may have these signs and symptoms that can be mild or serious:
- Coughing or wheezing
- Having lots of mucus in the lungs
- Many lung infections, such as pneumonia and bronchitis
- Shortness of breath
- Salty skin
- Slow growth, even with a big appetite
- Meconium ileus, when meconium gets stuck in a newborn’s intestine.
 Meconium is a baby's first bowel movement. It can be green, brown or black in color.
Meconium is a baby's first bowel movement. It can be green, brown or black in color. - Bowel movements that are frequent, loose, large or look greasy
- Stomach pain or bloating
If your baby has CF, how are lung and breathing problems treated?
Many lung infections in babies who have CF are caused by bacteria that don’t usually cause problems for healthy babies. If your baby has CF, medicines like antibiotics often cannot get rid of all the bacteria in their lungs. These infections can lead to lung damage.
Your child’s treatment depends on the kind of symptoms they have and how severe the symptoms are. Certain medicines can help children with CF breathe better and prevent infections. Some come as a mist that your child breathes into the lungs. Medicines used for CF include:
- Mucus-thinners. Medicines like dornase alfa (Pulmozyme®) help thin mucus, making it easier to cough out.
- Bronchodilators.
 These medicines help open the airways to clear mucus from the lungs. Albuterol (Proventil® and Ventolin®) is an example.
These medicines help open the airways to clear mucus from the lungs. Albuterol (Proventil® and Ventolin®) is an example. - Antibiotics. These are medicines that kill infections caused by bacteria. Tobramycin ( Tobi®) is a common inhaled antibiotic, and azithromycin is a common antibiotic taken by mouth.
- Ibuprofen. This medicine can help reduce lung redness and swelling that make breathing difficult.
- Hypertonic saline. Inhaling this salt-water mist helps draw more water into the airways. This helps thin the mucus.
Your child’s provider may recommend that they get lots of physical activity or that you use other therapies to vibrate (shake) the chest to help loosen mucus in her lungs. This can make it easier for your child to cough mucus up and out of the lungs.
If your child’s CF becomes life-threatening, a lung transplant may be an option. This is a major operation that is becoming more successful in treating CF.
If your baby has CF, how are growth and digestion problems treated?
Some children with CF gain weight and grow normally. But many grow more slowly than other children.
Most children with CF need to take special medicines that help their bodies get nutrients from food. This helps with weight gain and digestion.
To help them grow, children with CF need healthy, high-calorie meals. They need extra vitamins, especially vitamins A, D, E, and K. A dietitian with experience in treating children who have CF can help you create your child’s meal plan for a healthy weight gain. A dietician is a person who has special training in helping people eat healthy.
Some teens or young adults with CF may get CF-related diabetes. This is usually treated by getting shots of insulin at mealtimes. It’s important to keep diabetes under control so that it doesn’t cause more lung problems.
More information
- Cystic Fibrosis Foundation
See also: Genetic counseling
Last updated: May, 2021
Cystic Fibrosis (for Parents) - Nemours KidsHealth
What Is Cystic Fibrosis?
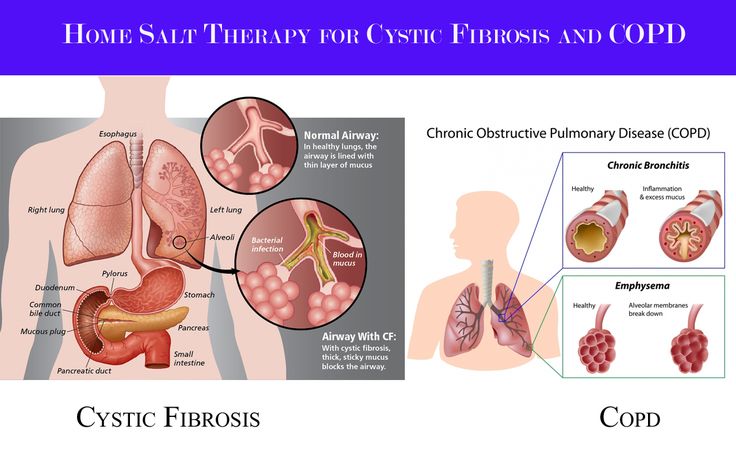
Cystic fibrosis (CF) is an inherited disease in which the body makes very thick, sticky mucus. The mucus causes problems in the lungs, pancreas, and other organs.
The mucus causes problems in the lungs, pancreas, and other organs.
People with cystic fibrosis (SIS-tik fye-BROH-sis) get lung infections often. Over time, they have more trouble breathing. They also have digestive problems that make it hard to gain weight.
What Are the Signs & Symptoms of Cystic Fibrosis?
CF can cause symptoms soon after a baby is born. The first sign a baby might have cystic fibrosis is an intestinal blockage called meconium ileus. Other kids don't have symptoms until later on. Cystic fibrosis can be mild or severe, depending on the person.
Symptoms of cystic fibrosis include:
- lung infections or pneumonia
- wheezing
- coughing with thick mucus
- bulky, greasy bowel movements
- constipation or diarrhea
- trouble gaining weight or poor height growth
- very salty sweat
Some kids also might have nasal polyps (small growths of tissue inside the nose), frequent sinus infections, and tiredness.
How Is Cystic Fibrosis Diagnosed?
Newborn screening tests catch most cases of CF. If the screening test is positive, or if a child has cystic fibrosis symptoms, doctors do a painless sweat test. They collect sweat from an area of skin (usually the forearm) to see how much chloride (a chemical in salt) is in it. People with CF have higher levels of chloride.
Most children with CF are diagnosed by the time they're 2 years old. But someone with a mild form may not be diagnosed until they are a teen.
How Is Cystic Fibrosis Treated?
Kids with CF will have it all their lives. Doctors use different medicines depending on a child's needs. But all people with CF need to:
- Loosen and clear mucus. There are different ways to do this. The doctor might recommend a child:
- get regular exercise
- use an inhaler or nebulizer
- do breathing exercises and cough on purpose
- wear a therapy vest that shakes the chest
- have chest physical therapy (when a parent or trained person bangs gently on the chest or back)
- Prevent or fight lung infections.
 Washing hands well and often, avoiding people who are sick, and staying at least 6 feet away from others with CF can help prevent infections. Taking preventive antibiotics also can help.
Washing hands well and often, avoiding people who are sick, and staying at least 6 feet away from others with CF can help prevent infections. Taking preventive antibiotics also can help. - Take enzymes. Most kids with CF need enzymes to help them digest food and get nutrients from it.
- Eat a high-calorie diet and take vitamin supplements, when needed.
What Causes Cystic Fibrosis?
Cystic fibrosis is caused by a change (mutation) in the gene that makes cystic fibrosis transmembrane regulator (CFTR) protein. To have CF, a baby must get two copies of the CF gene, one from each parent.
What Happens in Cystic Fibrosis?
In CF, the body makes abnormal CFTR protein or none at all. Without normal CFTR protein, the cells lining the pathways (tubes) inside some organs make thick, sticky mucus rather than the normal thin, watery kind.
Thick mucus can trap bacteria in the lungs, leading to infection, inflammation, and breathing problems.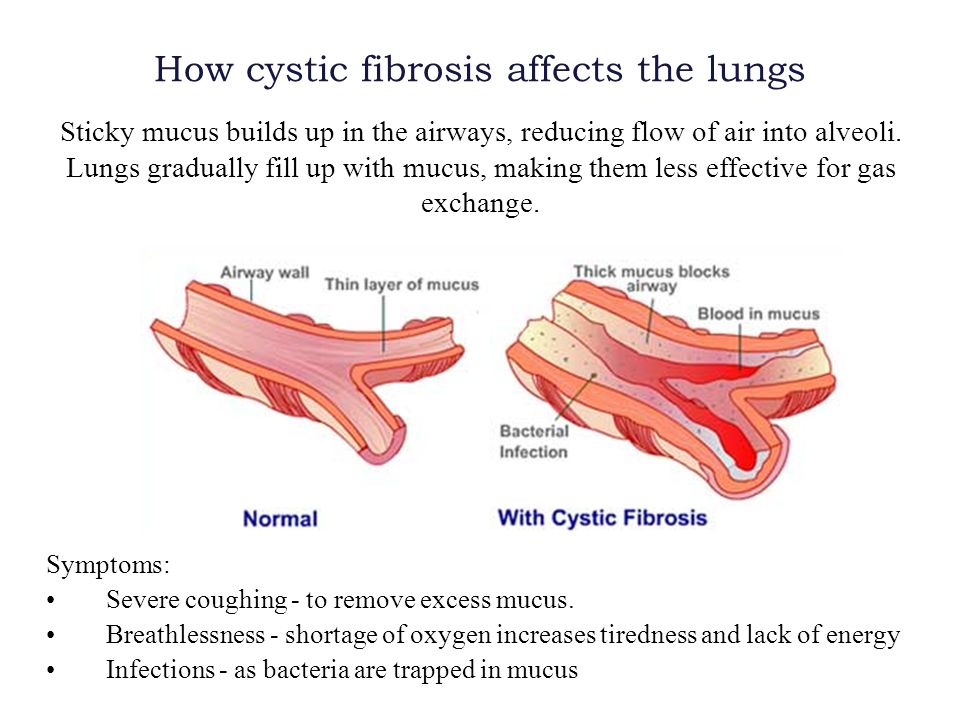 Mucus also can block the path where digestive enzymes flow between the pancreas and the intestines. This makes it hard for a child to digest food and get the vitamins and nutrients they need from it.
Mucus also can block the path where digestive enzymes flow between the pancreas and the intestines. This makes it hard for a child to digest food and get the vitamins and nutrients they need from it.
Thick mucus can also affect the liver, the sweat glands, and the reproductive organs.
How Can Parents Help?
To help your child:
- Follow the treatment plan. Help your child stay as healthy as possible. Give medicines as directed, serve high-calorie meals and snacks, and follow instructions for clearing chest mucus.
- Offer encouragement. Help your child find pastimes to enjoy, like art, music, reading, or learning to cook. It's important for kids with CF to get exercise, so also look for ways your child can stay physically active. Maybe you can do some of them together.
- Turn to the care team. Your child's care team can offer practical tips on living with CF, and information about clinical trials, support groups, and new therapies.
- Learn all you can about CF. Experts are always working on new treatments to help people with CF have a better quality of life and live longer. Online, turn to resources like the Cystic Fibrosis Foundation website.
- Teach self-care as your child gets older. Start early to help your child understand and manage CF. Encourage an older child or teen to handle some parts of their health care, like disinfecting equipment or asking questions at doctor visits. Ask the care team about ways you can help your child get ready for things like going to college or getting a job. Learning about cystic fibrosis and its care helps kids and teens become confident adults managing a chronic health condition.
Reviewed by: Larissa Hirsch, MD
Date reviewed: July 2020
Cystic fibrosis (cystic fibrosis) | Bebbo
Cystic fibrosis
Cystic fibrosis is a systemic hereditary disease characterized by damage to the external secretion glands and severe respiratory dysfunction.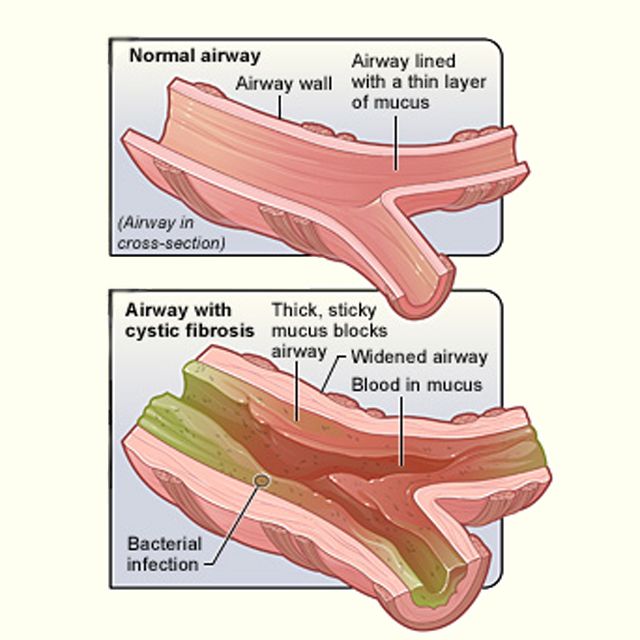 Cystic fibrosis produces thick, sticky mucus that blocks the lungs, clogs the airways, and makes it hard to cough up. Bacteria accumulate in these blockages, leading to many infections and damage to the lungs.
Cystic fibrosis produces thick, sticky mucus that blocks the lungs, clogs the airways, and makes it hard to cough up. Bacteria accumulate in these blockages, leading to many infections and damage to the lungs.
Children with cystic fibrosis cough a lot, have difficulty breathing, and get repeated respiratory tract infections. nine0005
In addition, thick mucus caused by cystic fibrosis can clog the pancreas, blocking the flow of pancreatic juices to the intestines for digestion. Because children with cystic fibrosis do not digest food properly, they may suffer from malnutrition, weight loss, and diarrhea.
Cystic fibrosis can lead to many long-term problems, such as recurring respiratory tract infections, poor weight gain, and diabetes.
Men diagnosed with cystic fibrosis may have difficulty conceiving children. nine0005
With advances in diagnosis and treatment, children with cystic fibrosis survive to adulthood, however, such a diagnosis means that their life expectancy may be shorter than that of other people.
Causes of cystic fibrosis
Cystic fibrosis is a genetic disorder caused by a mutation in a gene that controls how much salt and water gets in and out of the body's cells.
A baby can be born with cystic fibrosis if both parents carry the gene. Most carriers of the gene are healthy and do not show any symptoms. nine0005
Parents who already have a child with CF have a 25 percent chance of having another child with CF.
Diagnosis of cystic fibrosis
If someone in your family has been diagnosed with cystic fibrosis, seek medical genetic counseling and get tested for cystic fibrosis. The presence of the gene can be determined with a simple blood test.
If you are pregnant and there is a possibility that your unborn child will inherit the gene for cystic fibrosis, you may have additional tests during pregnancy to find out if your child has it. nine0005
Your child will be tested for cystic fibrosis if they have symptoms that make them suspicious, such as persistent respiratory and/or bowel problems. This is a simple test called a sweat test, which involves stimulating a small area of the skin to produce sweat.
This is a simple test called a sweat test, which involves stimulating a small area of the skin to produce sweat.
In some countries, newborns may be referred for testing for cystic fibrosis. Such an analysis includes taking blood from the heel of the baby. If the test results show high levels of an enzyme called immunoreactive trypsin (IRT), then a genetic test for common gene mutations can show if your child has cystic fibrosis. nine0005
Care and treatment for children with cystic fibrosis
There is no cure for cystic fibrosis.
If your child has cystic fibrosis, they will likely be treated by a specialized multidisciplinary team of cystic fibrosis specialists in a specialized unit.
Cystic fibrosis treatment can be intensive and time consuming. Children need to visit such a specialized unit regularly, and some will have to go to the hospital frequently for treatment of respiratory tract infections. nine0005
Physiotherapy and various treatments can help keep children healthy and improve their quality of life.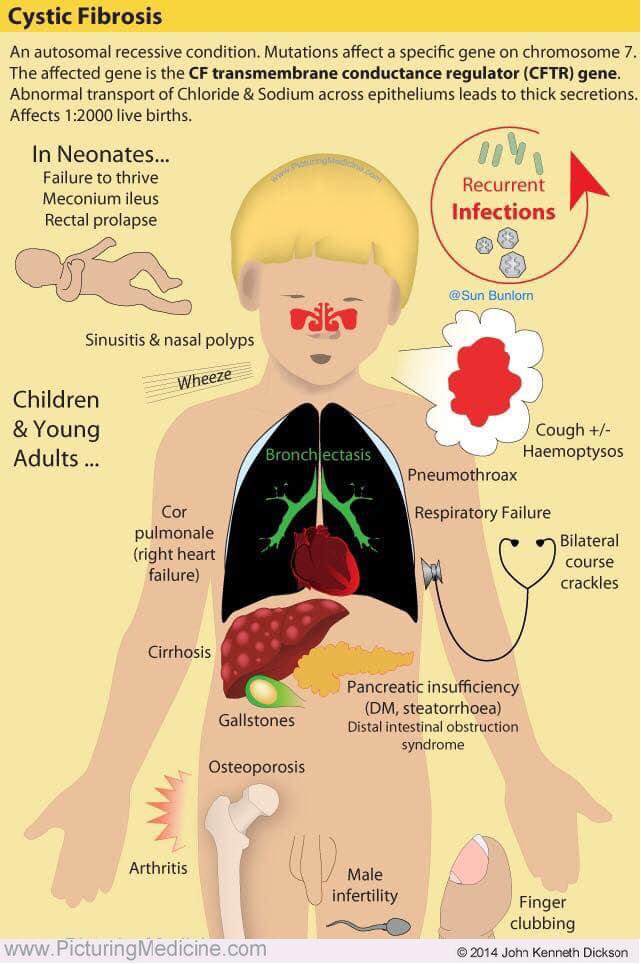 Children also require special lung-cleansing treatments that need to be done twice a day. These procedures are usually performed by the parent or caregiver of the child.
Children also require special lung-cleansing treatments that need to be done twice a day. These procedures are usually performed by the parent or caregiver of the child.
Usually several courses of daily antibiotics are needed. Children with cystic fibrosis may also need nutritional supplements and steroids to treat lung infections. Physicians sometimes recommend enzyme replacement therapy to help improve food absorption, as well as a high-energy diet with salt and vitamin supplements. nine0005
If your child has cystic fibrosis, you and your child may be seen by any or all of the following health professionals:
- genetic counselor
- pediatrician
- pulmonologist
- nutritionist
- physiotherapist
Living with cystic fibrosis
Keeping children with cystic fibrosis active, exercising and getting chest physiotherapy can help keep their lungs healthy. nine0005
A child with cystic fibrosis may miss school frequently due to the need for regular hospital stays.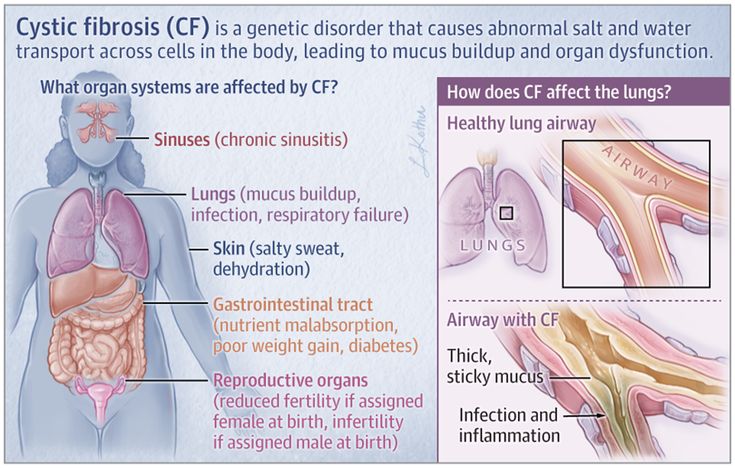 If your child has cystic fibrosis, it is important to tell teachers about the condition and communicate with them regularly.
If your child has cystic fibrosis, it is important to tell teachers about the condition and communicate with them regularly.
With good treatment, people with cystic fibrosis can live full lives well into middle age.
what it is, risk factors, symptoms, treatment
Cystic fibrosis or cystic fibrosis is a genetic disease that provokes damage to the external secretion glands, which leads to disruption of the respiratory and digestive organs. Men with cystic fibrosis are often infertile. nine0005
Contents
- Disease probability and statistics
- Diagnosis of cystic fibrosis
- How cystic fibrosis manifests itself
- Treatment
- Cystic fibrosis today
- Note
Cystic fibrosis is usually diagnosed in children under two years of age, most often in the first weeks of life.
The disease is caused by mutations in the CFTR (cystic fibrosis transmembrane regulator) gene.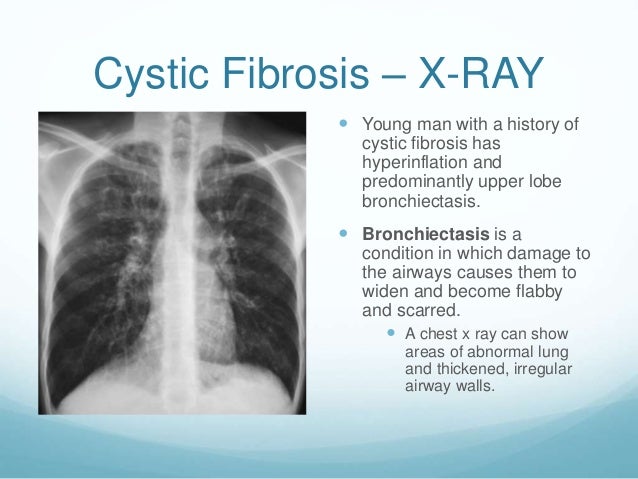
1000
new cases of cystic fibrosis are diagnosed in the world every year
CFTR is a protein that transports chloride ions across the membrane of cells that produce mucus, sweat, saliva, tears, and digestive enzymes. These ions are needed to control the movement of water, which liquefies the secretions of the glands. Due to mutations, the CFTR protein is not assembled correctly and cannot fully perform its function.
As a result, the transport of chlorine ions is disrupted, and consequently, the movement of water into and out of cells is impaired. The cells produce a thick and sticky secretion. And this can lead to the formation of cysts inside the organs. nine0005
Disease incidence and statistics
Both parents must be asymptomatic carriers of the disease in order for a child to receive two copies of the gene that produces the defective protein. If only one of the parents is such a carrier, then the child will not have cystic fibrosis, but it may also be a carrier.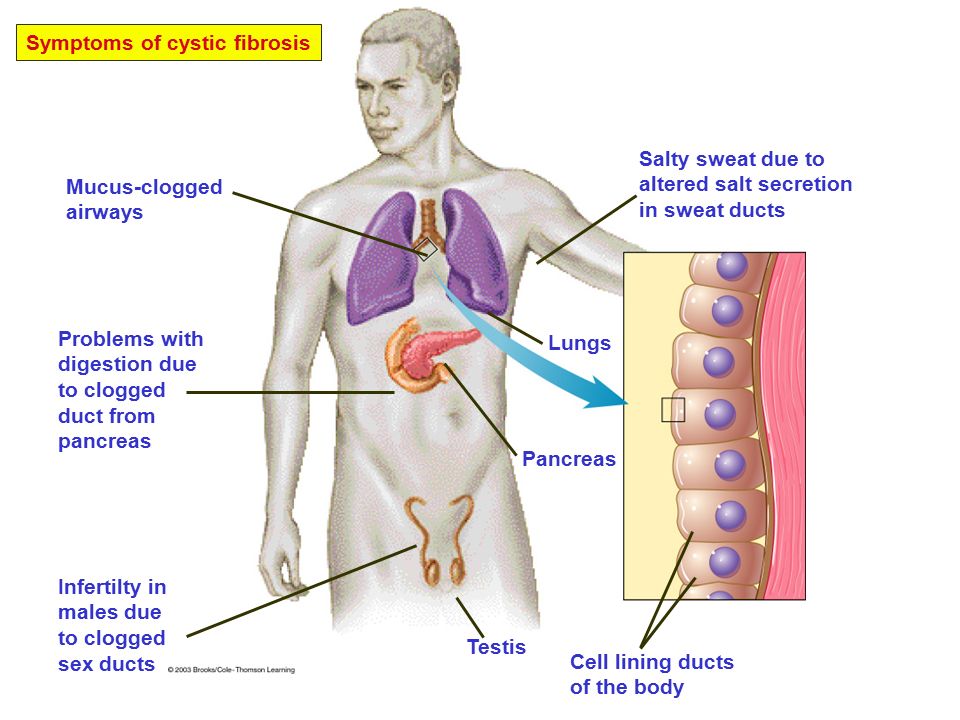 Most often, the disease occurs in people of European descent.
Most often, the disease occurs in people of European descent.
More than 70,000 people worldwide are now living with cystic fibrosis, with more than 1,000 new cases diagnosed each year. According to the registry of patients with cystic fibrosis in Russia in 2017, there were about 3,400 people with this diagnosis. nine0005
Probability of having a child with cystic fibrosis in carriers of mutated CFTR:
1. Both parents are carriers:
- 25% - children will have cystic fibrosis;
- 50% - children will be carriers;
- 25% - Children will neither be carriers nor become sick.
2. One parent is a carrier, the other has cystic fibrosis:
- 50% - children will be carriers;
- 50% - children will have cystic fibrosis. nine0061
- 100% - children will be carriers of cystic fibrosis.
- 50% - children will be healthy;
- 50% - children will be carriers.
- persistent cough, sometimes with phlegm;
- frequent lung infections, including pneumonia and bronchitis;
- wheezing and shortness of breath;
- changes on the radiograph of the lungs;
- difficulty gaining height and weight with good appetite;
- frequent, loose, foul-smelling stools or difficult bowel movements.
- nutrition normalization;
- replacement therapy for pancreatic insufficiency;
- clearing the lungs of mucus;
- physiotherapy exercises;
- prescription of antibiotics in case of infection.
 nine0052
nine0052 - Cystic fibrosis is a genetic disease that causes damage to the external secretion glands, which leads to disruption of the respiratory and digestive organs.
- The disease is usually diagnosed before 2 years of age, more often in the first weeks of life through screening. nine0052
- If earlier many patients died in childhood, now life expectancy with cystic fibrosis reaches 50 years or more.
- Cystic fibrosis is incurable, but the course of the disease can be alleviated with lifelong daily treatments.
One parent is healthy, the other parent has cystic fibrosis:
One parent is healthy, the second is a carrier:
To find out the likelihood of illness in their future children, both parents should take a test for hereditary diseases.
Diagnosis of cystic fibrosis
In some countries where cystic fibrosis is common, newborn screening is done in the hospital. This helps to start treatment in a timely manner.
At the first stage, the content of immunoreactive trypsinogen (IRT) is evaluated. It is a precursor of a pancreatic enzyme that enters the child's bloodstream due to damage to the pancreas in cystic fibrosis. If the result is positive, the test is repeated on the 21st-28th day of life.
Cystic fibrosis is not the only reason RTIs can be raised. This can happen, for example, if the baby is premature or due to meconium contamination of the sample (most likely). Therefore, for accurate diagnosis, a “sweat test” is prescribed - sweat samples are taken from the child to check the concentration of chlorides in it (with cystic fibrosis, their level is increased).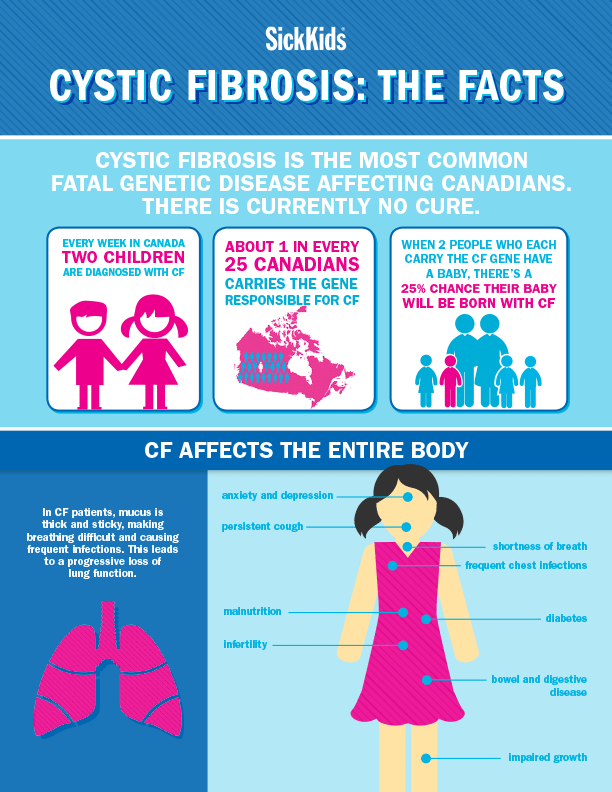 With a borderline result of a sweat test, additional examination methods are prescribed. nine0005
With a borderline result of a sweat test, additional examination methods are prescribed. nine0005
How cystic fibrosis manifests itself
Main symptoms:
Neonatal
Meconium (original feces) becomes thick and sticky, therefore accumulates in the intestines and is not excreted, which requires urgent surgical intervention.
In older children
In early childhood, the most obvious manifestation of the disease is pancreatic insufficiency. The secret clogs the ducts, so the intestines do not receive digestive enzymes in the right amount. The absorption of proteins, fats and fat-soluble vitamins is impaired, which is why the child does not gain weight well with a good appetite. nine0005
nine0005
Excess enzymes destroy cells leading to pancreatitis with development of cysts and fibrosis. Destruction of pancreatic tissue can disrupt its endocrine function and cause diabetes mellitus.
Pulmonary manifestations usually occur a little later. Due to the disease, the secret of the bronchi becomes viscous, sputum stagnates and often becomes infected, which leads to the development of pulmonary diseases.
Treatment
If a child is diagnosed with cystic fibrosis, the first thing parents can do is to work closely with the doctor as soon as the test results are available. nine0005
50 years
estimated median survival age for people with cystic fibrosis
It is important to understand that a lifelong treatment regimen is required - adherence to procedures literally every day.
Main recommendations:
Since 2019, a number of pharmaceutical companies have reduced the supply of important antibiotics to fight cystic fibrosis in Russia. Instead, doctors use domestically produced generics, the active substance of which is less effective. Because of this, courses of antibiotics have to be carried out more often, which exacerbates side effects - for example, allergies. It is possible to achieve individual purchases of foreign-made drugs if the medical commission recognizes side effects from the use of generics.
For some genetic mutations, the drug Ivacoftor is indicated.
The Whole Genome test can be used to assess the individual characteristics of the use of Ivacoftor.
Cystic Fibrosis Today
In 1955, the Cystic Fibrosis Foundation was founded, which sees as its goal a detailed study of the disease and alleviation of the lives of thousands of people.
The activities of this and other scientific foundations have borne fruit - in the past, mortality was mainly among children and adolescents.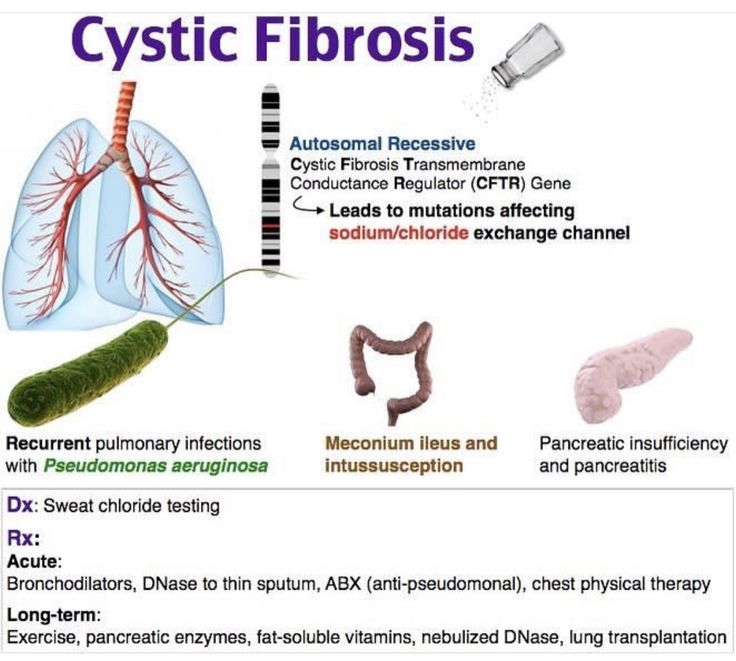 More than half of the patients died in infancy. nine0005
More than half of the patients died in infancy. nine0005
Adult cystic fibrosis patients now outnumber children in developed countries, and the estimated median survival age is approaching 50 years.
Note
To understand whether there is a risk of a disease in your family, we advise both parents to take the Atlas Genetic Test during pregnancy planning. It contains information on 322 hereditary conditions, including cystic fibrosis.