Testing for fragile x in pregnancy
FAQ: Carrier Testing for Fragile X Syndrome | Patient Education
What is fragile X syndrome?
Fragile X syndrome is the most common cause of inherited mental retardation. The condition affects approximately 1 in 3,600 males and 1 in 6,000 females.
Fragile X syndrome causes a range of symptoms. Abilities range from mild learning disabilities to severe mental retardation. Behavioral characteristics include autism, hyperactivity, short attention span and poor eye contact.
Physical features, such as a long face, large or prominent ears, flat feet and enlarged testes, are usually more noticeable in young adults than in children, and in males more than females.
How is fragile X syndrome inherited?
Fragile X syndrome is caused by a change in the fragile X mental retardation (FMR1) gene. The FMR1 gene is located on the X chromosome. This abnormal gene, which can be passed from generation to generation, is usually inherited through the gene that is carried by women.
A fragile X carrier is someone who has an altered FMR1 gene, but does not show any obvious signs or symptoms of fragile X syndrome. Women who are fragile X carriers have up to a 50 percent chance of having a child with fragile X syndrome. Men who are fragile X carriers will pass the altered gene to all of their daughters but none of their sons. Daughters of carrier men are expected to be intellectually normal but are at risk of having children affected with fragile X syndrome.
The genetics of fragile X syndrome are complicated. Genetic counseling is recommended when someone has a family history of fragile X syndrome or is shown to be a carrier of fragile X.
Who can be a fragile X carrier?
Anyone can be a carrier of fragile X syndrome. It is found among all ethnic backgrounds and racial groups. Approximately 1 in 250 women in the general population are carriers of the abnormal gene that causes fragile X syndrome. A woman of any age can have a child with fragile X syndrome, whether or not she has had previous children.
Who is at greater risk to be a fragile X carrier?
You are at greater risk if you have:
- A family history of fragile X syndrome
- A family history of mental retardation, developmental delay or autism of unknown cause
- Infertility problems associated with elevated follicle stimulating hormone (FSH) levels or premature ovarian failure (POF)
- A family history of adult onset ataxia and/or tremors
What is the fragile X carrier test and what do the results show?
The fragile X carrier test provides specific information about whether or not individuals are fragile X carriers, and about their risks of having a child with fragile X syndrome. The test is performed on a small sample of blood. Results are usually available within two weeks. Testing provides accurate results more than 99 percent of the time. Other causes of mental retardation are not identified through this test.
What are the possible results from a fragile X carrier test?
There are four possible results from a fragile X carrier test:
- Negative You are not a carrier for the most common alteration in the FMR1 gene and your baby is not at increased risk for fragile X syndrome.
- Intermediate Your results fall in the range between negative and premutation. Your baby is not at increased risk for fragile X syndrome. Future generations may be at risk for fragile X syndrome.
- Premutation You are a carrier for the altered FMR1 gene. You may be at risk for early menopause. Your baby is at risk for fragile X syndrome. Prenatal diagnostic testing is available.
- Full mutation You are a carrier for the altered FMR1 gene. Your baby is at risk for fragile X syndrome. Prenatal diagnostic testing is available.
Genetic counseling is recommended and available when someone is shown to be a carrier of fragile X.
Is prenatal testing available?
Prenatal testing can be performed by amniocentesis at 16 to 20 weeks or by chorionic villus sampling (CVS) at 10 to 13 weeks to determine if a fetus has inherited the fragile X gene.
For more information about fragile X syndrome, genetic counseling, or to arrange carrier testing, please contact the Prenatal Diagnostic Center.
Prenatal Screening for Fragile X: Carriers, Controversies, and Counseling
Rev Obstet Gynecol. 2013; 6(1): e1–e7.
, BS,1, MD, FACOG, FACMG,2 and , MD, FACOG, FACMG2
Author information Copyright and License information Disclaimer
In addition to causing developmental disability in future offspring, fragile X carrier status has important reproductive and mental health implications for the individual being tested. Accordingly, prenatal carrier screening and diagnosis using DNA-based molecular methods has become crucial in early detection, intervention, and family planning. Although the list of known genetic disorders is growing daily, controversy remains over who should be tested for fragile X. FMR1 gene mutations can result in inherited intellectual disability, infertility, and neurodegeneration syndromes that are encountered by clinicians in a variety of settings. Patients and clinicians are still largely unfamiliar with this disorder, its complicated inheritance, and its heterogeneous phenotype. Debate continues over who should be offered prenatal carrier screening. As more disease screening is offered, pretest counseling will become only more complex and clinicians will further struggle to balance the needs of the individual and allocation of public health resources.
Debate continues over who should be offered prenatal carrier screening. As more disease screening is offered, pretest counseling will become only more complex and clinicians will further struggle to balance the needs of the individual and allocation of public health resources.
Key Words: Fragile X syndrome, FMR1 gene mutation, Prenatal screening, Genetic counseling
Unlike most prenatal carrier testing, Fragile X carrier status has important reproductive and health implications both for the individual being tested as well as potential risks for developmental disability in future offspring. Clinicians in a variety of settings encounter the infertility, intellectual disability and neurodegenerative syndromes resulting from FMR1 mutations. Accordingly, physicians must be comfortable with patient counseling regarding Fragile X and should remain vigilant in identifying patients who have indications for prenatal screening. Below we review the complicated inheritance of Fragile X and its varied phenotype. The current guidelines for prenatal screening are described and common counseling issues are addressed. Finally, we discuss universal prenatal carrier screening, a topic which will become only more complex as clinicians further struggle to balance the needs of the individual and allocation of public health resources.
The current guidelines for prenatal screening are described and common counseling issues are addressed. Finally, we discuss universal prenatal carrier screening, a topic which will become only more complex as clinicians further struggle to balance the needs of the individual and allocation of public health resources.
Case 1
DM is a 33-year-old white woman who presents to your clinic during her first pregnancy for a first trimester screen for fetal aneuploidy. You take an extended history to determine her risk for a variety of genetic carrier screening tests available to her. She tells you that her maternal grandmother died of breast cancer at age 73. Her maternal grandfather lived to be age 75, but was severely demented toward the end of his life. DM’s parents are living and healthy. Her older sister is healthy but has a son with mental retardation. Her younger brother is age 21 and healthy with no children.
Case 2
JP is a 32-year-old newlywed who comes to your clinic for preconception counseling. She is concerned that she may have some trouble getting pregnant as infertility seems to “run in her family.” JP’s older sister began to have hot flashes at age 34. Her older sister required in vitro fertilization to conceive her niece, who is healthy. Shortly after having her only daughter at age 37, her sister was told that she had gone through menopause. JP wants to know if there is any way to predict if she will have similar problems. The remainder of JP’s family history is unremarkable for infertility or mental retardation.
She is concerned that she may have some trouble getting pregnant as infertility seems to “run in her family.” JP’s older sister began to have hot flashes at age 34. Her older sister required in vitro fertilization to conceive her niece, who is healthy. Shortly after having her only daughter at age 37, her sister was told that she had gone through menopause. JP wants to know if there is any way to predict if she will have similar problems. The remainder of JP’s family history is unremarkable for infertility or mental retardation.
Case 3
RG is a 42-year-old woman who presents to your reproductive endocrinology clinic with her partner for in vitro fertilization. She is a college biology professor and has read a lot about the process. An unrelated family friend has donated her eggs for the couple’s use. RG would like to learn as much as possible about the pregnancy and asks you what genetic tests will be run on the donor eggs prior to implantation.
Fragile X syndrome is the most common inherited form of intellectual and developmental disability worldwide. It has an estimated prevalence of 1 in 3600 males, and 1 in 4000 to 6000 females in various ethnic groups. Unlike other causes of mental retardation such as Down syndrome, phenotypic features of fragile X often are not apparent until later in childhood, making it difficult to diagnose during the newborn period. The mean age of diagnosis is age 3 years, and one study found that approximately one-quarter of families had a second child with the full mutation before the first child was diagnosed.1 In addition to causing developmental disability in future offspring, fragile X carrier status has important reproductive and mental health implications for the individual being tested. Accordingly, prenatal carrier screening and diagnosis using DNA-based molecular methods has become crucial in early detection, intervention, and family planning. Although the list of known genetic disorders is growing daily, controversy remains over who should be tested for fragile X.
It has an estimated prevalence of 1 in 3600 males, and 1 in 4000 to 6000 females in various ethnic groups. Unlike other causes of mental retardation such as Down syndrome, phenotypic features of fragile X often are not apparent until later in childhood, making it difficult to diagnose during the newborn period. The mean age of diagnosis is age 3 years, and one study found that approximately one-quarter of families had a second child with the full mutation before the first child was diagnosed.1 In addition to causing developmental disability in future offspring, fragile X carrier status has important reproductive and mental health implications for the individual being tested. Accordingly, prenatal carrier screening and diagnosis using DNA-based molecular methods has become crucial in early detection, intervention, and family planning. Although the list of known genetic disorders is growing daily, controversy remains over who should be tested for fragile X.
The molecular genetics of fragile X syndrome are uniquely complex. Fragile X syndrome is an X-linked, dominant disorder caused by the expansion of an unstable cysteine-guanine-guanine (CGG) trinucleotide repeat on the 5′ untranslated region of the fragile X mental retardation-1 gene (FMR1). Mutations are categorized into four allelic forms based on repeat size: unaffected (< 45 repeats), intermediate (45–54 repeats), premutation (55–200 repeats), and full mutation (> 200 repeats).2
Fragile X syndrome is an X-linked, dominant disorder caused by the expansion of an unstable cysteine-guanine-guanine (CGG) trinucleotide repeat on the 5′ untranslated region of the fragile X mental retardation-1 gene (FMR1). Mutations are categorized into four allelic forms based on repeat size: unaffected (< 45 repeats), intermediate (45–54 repeats), premutation (55–200 repeats), and full mutation (> 200 repeats).2
The large repeat length in the fragile X full mutation results in methylation and subsequent inactivation of the FMR1 gene. This results in decreased encoding of the fragile X mental retardation protein, which may be important for protein synthesis regulation at the level of the dendritic spine.3 Males with the full mutation all exhibit clinical features of fragile X syndrome. Females with the full mutation may or may not exhibit features of fragile X syndrome, depending on the degree of X inactivation. X inactivation is the random silencing of one of the X chromosomes in the cells of all female mammals. If the mutated chromosome is inactivated, its gene product is not produced and the resulting phenotype is less severe.
If the mutated chromosome is inactivated, its gene product is not produced and the resulting phenotype is less severe.
In addition, the inheritance pattern of fragile X is distinctive because unstable alleles may undergo mitotic expansion when transmitted from female carriers to offspring (). In this way, premutation carriers, who themselves display no cognitive impairment and may have no family history of cognitive impairment, may have offspring with the full mutation and therefore fragile X syndrome. The prevalence of premutation carriers in the United States is approximately 1 in 86 women with a family history of mental retardation and 1 in 257 women without any known risk factors for fragile X syndrome.4 Moreover, premutation carriers may also suffer from fragile X-associated premature ovarian insufficiency and tremor/ataxia syndrome. It is thought that in premutation carriers, FMR1 mRNA is produced in toxic amounts in leukocytes and neurons, resulting in these disorders. 5
5
Table 1
Maternal Allele Classification and Expansion to Full Mutation
| Maternal Mutation Status | Maternal CGG Repeat Size | Full Mutation Expansion (%) in Next Generation |
|---|---|---|
| Unaffected | < 45 | 0 |
| Intermediate | 45–54 | 0 |
| Premutation | 55–59 | < 2 |
| 60–69 | 2 | |
| 70–79 | 32 | |
| 80–89 | 74 | |
| 90–99 | 94 | |
| 100–200 | 98 | |
| Full Mutation | > 200 |
Open in a separate window
Data from Nolin SL et al. 33
33
Intermediate mutations have not been shown to expand to a full mutation in one generation. Still, alleles of this size may be associated with fragile X syndrome in future generations or distant relatives.6
Phenotype
Males with the full fragile X mutation present with intellectual impairment ranging from learning disability to mental retardation, autism, and severe behavioral difficulties (). Males also display characteristic physical features, such as long, narrow faces, prominent ears, connective tissue laxity, muscle hypotonia, and macro-orchidism. The prevalence and severity of fragile X syndrome in females is attenuated because of X-inactivation. Still, approximately 50% of females with the fragile X full mutation will show some features of the disorder.
Table 2
Clinical Features of FMR1-related Disorders
| Fragile X syndrome | Large occipitofrontal head circumference |
| Long face, prominent jaw, prominent ears | |
| Macroorchidism | |
| Connective tissue laxity, mitral valve prolapse | |
| Hypotonia | |
| Intellectual disability, low IQ, speech and motor delay | |
| Behavioral problems, autism and attention deficit spectrum disorders | |
| Fragile X-associated premature ovarian insufficiency | Premature ovarian failure prior to age 40 |
| Impaired ovarian response | |
| Fragile X-associated tremor/ataxia syndrome | Intention tremor |
| Cerebellar gait ataxia | |
| Dementia |
Open in a separate window
Fragile X is the most commonly identified cause of autism, a pervasive developmental disorder characterized by impaired social interaction, communication, and restricted, stereotyped behaviors. Studies estimate that 5% of patients with autism have fragile X.7 Twenty-five percent of boys with fragile X and 6% of girls with fragile X meet the diagnostic criteria for autism.8,9 In addition, the majority of patients with fragile X will display some autistic symptoms such as poor eye contact, hand flapping, and speech perseveration. Lower levels of fragile X mental retardation protein have been correlated with higher scores on neuropsychological measures of autism.9 Current guidelines suggest that all children with autism be offered genetic counseling and testing for fragile X syndrome.10 Some studies have proposed, however, that children with fragile X will demonstrate a unique profile of autism symptoms when compared with children with idiopathic autism.11
Studies estimate that 5% of patients with autism have fragile X.7 Twenty-five percent of boys with fragile X and 6% of girls with fragile X meet the diagnostic criteria for autism.8,9 In addition, the majority of patients with fragile X will display some autistic symptoms such as poor eye contact, hand flapping, and speech perseveration. Lower levels of fragile X mental retardation protein have been correlated with higher scores on neuropsychological measures of autism.9 Current guidelines suggest that all children with autism be offered genetic counseling and testing for fragile X syndrome.10 Some studies have proposed, however, that children with fragile X will demonstrate a unique profile of autism symptoms when compared with children with idiopathic autism.11
Women who are carriers of the fragile X premutation are not only at risk of transmitting the fragile X full mutation to their offspring, but are also at increased risk of developing primary ovarian insufficiency. Schwartz and colleagues conducted the first study to investigate the gynecologic histories of premutation and full mutation carriers in 1994, showing that premutation carriers had higher rates of irregular menses and use of hormones to aid fertility than full mutation carriers and noncarriers.12 The phrase fragile-X-associated premature ovarian insufficiency (FXPOI) encompasses the variety of ovarian dysfunction observed in premutation carriers. Approximately 20% of carriers with the fragile X premutation experience overt ovarian failure (absent menses and elevated follicle-stimulating hormone prior to age 40) compared with 1% of the general population. In addition, premutation carriers experience menopause, on average, 5 years earlier than the general population. Two percent of patients seen in fertility clinics with premature ovarian failure are found to have the fragile X premutation-much higher than rates in the general population.13 Of note, recent research in fertility clinics has also shown an increased association between occult primary ovarian insufficiency (regular menses but impaired ovarian response) and the fragile X premutation.
Schwartz and colleagues conducted the first study to investigate the gynecologic histories of premutation and full mutation carriers in 1994, showing that premutation carriers had higher rates of irregular menses and use of hormones to aid fertility than full mutation carriers and noncarriers.12 The phrase fragile-X-associated premature ovarian insufficiency (FXPOI) encompasses the variety of ovarian dysfunction observed in premutation carriers. Approximately 20% of carriers with the fragile X premutation experience overt ovarian failure (absent menses and elevated follicle-stimulating hormone prior to age 40) compared with 1% of the general population. In addition, premutation carriers experience menopause, on average, 5 years earlier than the general population. Two percent of patients seen in fertility clinics with premature ovarian failure are found to have the fragile X premutation-much higher than rates in the general population.13 Of note, recent research in fertility clinics has also shown an increased association between occult primary ovarian insufficiency (regular menses but impaired ovarian response) and the fragile X premutation. 14 Therefore, the fragile X premutation is likely to be an important cause of decreased fecundity that should be considered in reproductive endocrinology clinics.
14 Therefore, the fragile X premutation is likely to be an important cause of decreased fecundity that should be considered in reproductive endocrinology clinics.
Premutation carriers may also suffer from a neurodegenerative disorder known as fragile X-associated tremor/ataxia syndrome (FXTAS) later in life. Onset is typically in the sixth decade of life and is thought to affect approximately 40% of male premutation carriers and 80% of female carriers over age 50.15 Currently, most patients with FXTAS are only identified after a grandchild is diagnosed with fragile X syndrome despite the fact that FXTAS is the most common known single-gene cause of tremor/ataxia in the elderly. This is likely because of decreased familiarity of many physicians with this disorder and the heterogeneity with which it presents. The presenting symptom is often an isolated intention tremor progressing to cerebellar gait ataxia, frontal executive dysfunction, and psychiatric derangements. 16 Magnetic resonance imaging in such patients displays global brain atrophy and white matter lesions of the middle cerebellar peduncles.17 FXTAS is a progressive disorder with a life expectancy of 5 to 20 years from time of diagnosis. At the end stage, patients are often bedridden, demented, dysarthric, and incontinent.
16 Magnetic resonance imaging in such patients displays global brain atrophy and white matter lesions of the middle cerebellar peduncles.17 FXTAS is a progressive disorder with a life expectancy of 5 to 20 years from time of diagnosis. At the end stage, patients are often bedridden, demented, dysarthric, and incontinent.
Current guidelines from the American Congress of Obstetricians and Gynecologists (ACOG) recommend prenatal screening and genetic counseling for fragile X in women with a family or personal history of fragile X, unexplained mental retardation or developmental delay, or premature ovarian insufficiency ().18 In addition, ACOG recommends that all women who request fragile X carrier screening be offered DNA testing after appropriate genetic counseling.18 The American College of Medical Genetics (ACMG) adds that DNA analysis for fragile X syndrome should be included as part of the diagnostic evaluation in patients with isolated cognitive impairment or cerebellar ataxia and intention tremors, especially if they are male. 10
10
Table 3
Current ACOG and ACMG Guidelines
| Who Should Be Offered Screening? | |
|---|---|
| Any patient with a personal or family history of … | • Fragile X or fragile X-related disorders |
| • Unexplained mental retardation or developmental delay | |
| • Autism | |
| • Ovarian insufficiency or elevated FSH at age < 40 years of unknown cause | |
| Men with… | • Isolated cerebellar ataxia with tremor |
| • Isolated cognitive impairment | |
| Any woman who requests fragile X carrier screening regardless of history | |
Open in a separate window
ACMG, American College of Medical Genetics; ACOG, American Congress of Obstetricians and Gynecologists;
FSH, follicle-stimulating hormone
Prenatal carrier screening programs in women who do not have the above indications for testing have been debated for a variety of reasons.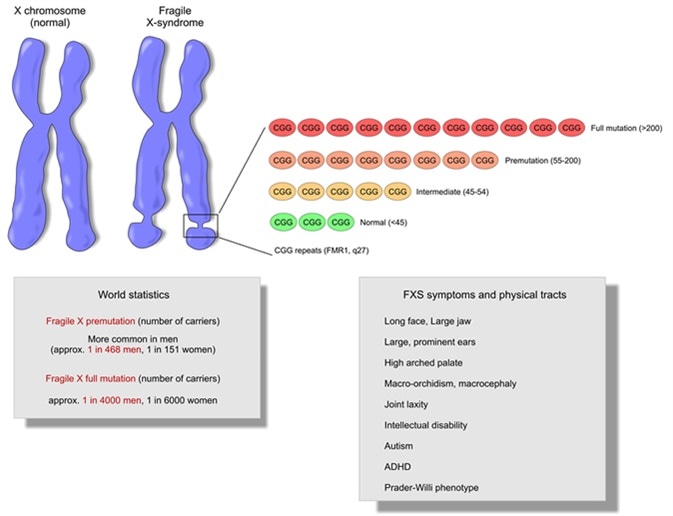 First, fragile X has clinically significant adverse consequences on affected individuals and their families comparable to Down syndrome.19 The incidence of fragile X is similar to genetic diseases for which we already offer routine screening, such as cystic fibrosis.20 Furthermore, adherence to current screening guidelines may only identify about 50% of fragile X carriers.21 A recent survey has shown that a majority of genetic health professionals supported prenatal fragile X screening programs.22
First, fragile X has clinically significant adverse consequences on affected individuals and their families comparable to Down syndrome.19 The incidence of fragile X is similar to genetic diseases for which we already offer routine screening, such as cystic fibrosis.20 Furthermore, adherence to current screening guidelines may only identify about 50% of fragile X carriers.21 A recent survey has shown that a majority of genetic health professionals supported prenatal fragile X screening programs.22
Fragile X prenatal screening serves a dual purpose: it allows patients to make educated reproductive decisions and it gives them information about their personal health. Accurate prenatal fetal diagnosis is available and the choice can be offered whether to continue pregnancy. Parents of children with fragile X have expressed the desire for prenatal screening because it would lead to earlier diagnosis, interventions, and altered family planning.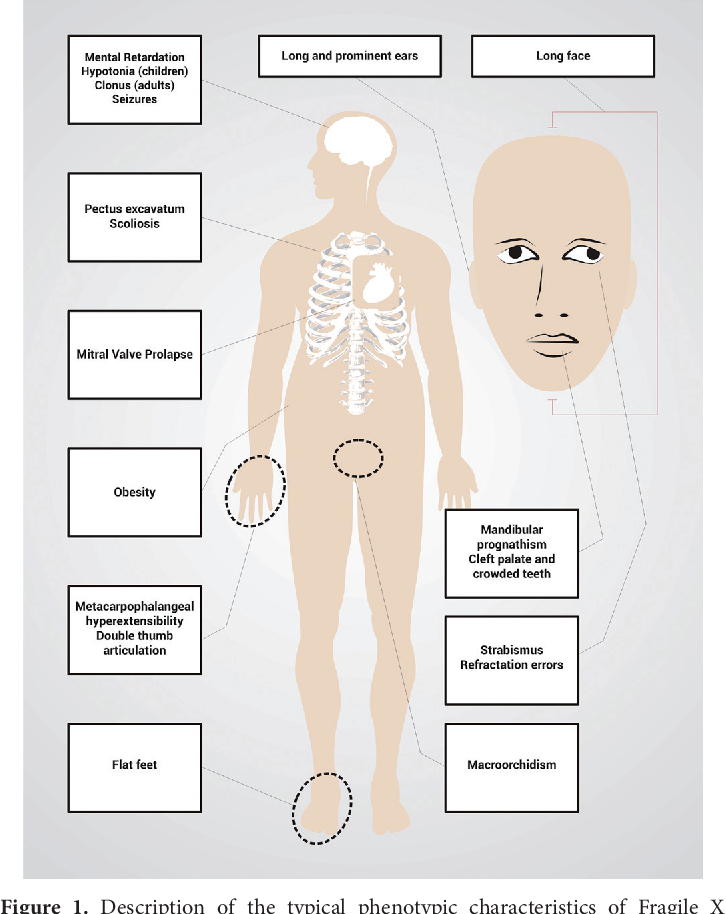 23,24 Similarly, identification of carriers at risk for premature ovarian failure may allow for earlier and more effective reproductive interventions in those desiring pregnancy. Finally, it has been shown that screening all women prenatally, regardless of family history or specific risk factors, is cost effective when considering the burden of raising a child with fragile X syndrome.25,26
23,24 Similarly, identification of carriers at risk for premature ovarian failure may allow for earlier and more effective reproductive interventions in those desiring pregnancy. Finally, it has been shown that screening all women prenatally, regardless of family history or specific risk factors, is cost effective when considering the burden of raising a child with fragile X syndrome.25,26
Highly accurate prenatal diagnosis of fetuses at risk for fragile X is currently available via chorionic villus sampling (CVS) and amniocentesis. Expansion and methylation status of prenatal samples can be determined using a combination of polymerase chain reaction and southern analysis. Methylation results must be interpreted with caution from CVS and patients should be aware that a follow-up amniocentesis may be required if testing yields an ambiguous result. Information about gender acquired at the time of sampling can aid patients and clinicians in approximating phenotypic severity, with female fetuses typically having milder disease.
All pregnant women who are full mutation or premutation carriers should be offered prenatal diagnosis.27 Recommendations for prenatal diagnosis for intermediate carriers are difficult to make and must be made on a case-by-case basis. On one hand, prenatal diagnosis gives information about allele instability and potential health risks for future offspring. For example, an intermediate carrier could discover that her daughter is a premutation carrier, prompting early discussion about reproductive choices when that daughter is older. Particularly anxious women may also find reassurance in fetal diagnosis, putting them at ease for the remainder of their pregnancy. On the other hand, prenatal diagnosis may cause undue angst in intermediate carriers when there is no demonstrated risk of expansion to the full mutation in one generation.
The identification of cell-free fetal DNA and RNA in maternal circulation holds promise for future development of noninvasive fragile X prenatal diagnosis. Cell-free fetal DNA testing is commercially available worldwide to determine gender, aneuploidy, and fetal RhD blood type.28 Monogenic testing is in development.29 Although cellfree fetal DNA is safe for the fetus, its cost and lower sensitivity and specificity have prevented it from replacing older techniques.30
Cell-free fetal DNA testing is commercially available worldwide to determine gender, aneuploidy, and fetal RhD blood type.28 Monogenic testing is in development.29 Although cellfree fetal DNA is safe for the fetus, its cost and lower sensitivity and specificity have prevented it from replacing older techniques.30
Fragile X carrier testing raises distinctly complicated genetic counseling issues. First, an extensive multigenerational family history must be taken to assess whether screening would be beneficial to the patient. When physicians consider FMR1 testing, this history must include developmental, neurodegenerative, and reproductive disability across multiple generations.31 Patients are generally unaware of fragile X prior to counseling, and, as such, the complexity of the fragile X inheritance pattern may be overwhelming to them. Physicians and counselors must educate patients about the varied outcomes possible and the implications of detecting full allele, premutation allele, and intermediate allele carrier results. Ideally, women should have sufficient time to review and consider the information given to them about carrier testing and reflect on their own psychosocial situation prior to making a decision about testing.
Ideally, women should have sufficient time to review and consider the information given to them about carrier testing and reflect on their own psychosocial situation prior to making a decision about testing.
Premutation carriers identified in the prenatal setting should be offered fetal diagnostic testing. New evidence suggests that the number of AGG interruptions may predict the risk of expansion of alleles with 45 to 69 repeats in the next generation.32 Currently, however, it is very difficult to accurately estimate an individual’s risk for FMR1 intermediate and premutation allele expansion. Of note, it was observed that women who carry newly identified fragile X premutation alleles and have no family history of fragile X have dramatically lower expansion rates to full mutations than patients with a positive family history.33 This result may be comforting to some patients. Women identified as carriers must be counseled about the risk for ovarian dysfunction and the reproductive options that are available to them. Family dynamics must also be considered and advice should be offered regarding testing other at-risk family members.
Family dynamics must also be considered and advice should be offered regarding testing other at-risk family members.
Intermediate carrier results may provoke worry because the risk for expansion, although very low, is uncertain. In addition, intermediate carriers should be notified that fragile X testing should be offered to future generations to determine allele stability and identify those at risk for offspring with the full mutation.
Prenatal identification of female fetuses with the fragile X full mutation also poses a significant challenge. X inactivation modifies the clinical phenotype in females so that the cognitive impairment is typically less severe than in males. Yet approximately 50% of females with the full mutation are affected, and phenotypic severity cannot be predicted prenatally.34 Uncertainty about the resulting phenotype generates significant anxiety and parents have a difficult choice to make.
Concerns regarding sufficient patient counseling and education are some of the greatest challenges to the implementation of widespread carrier screening.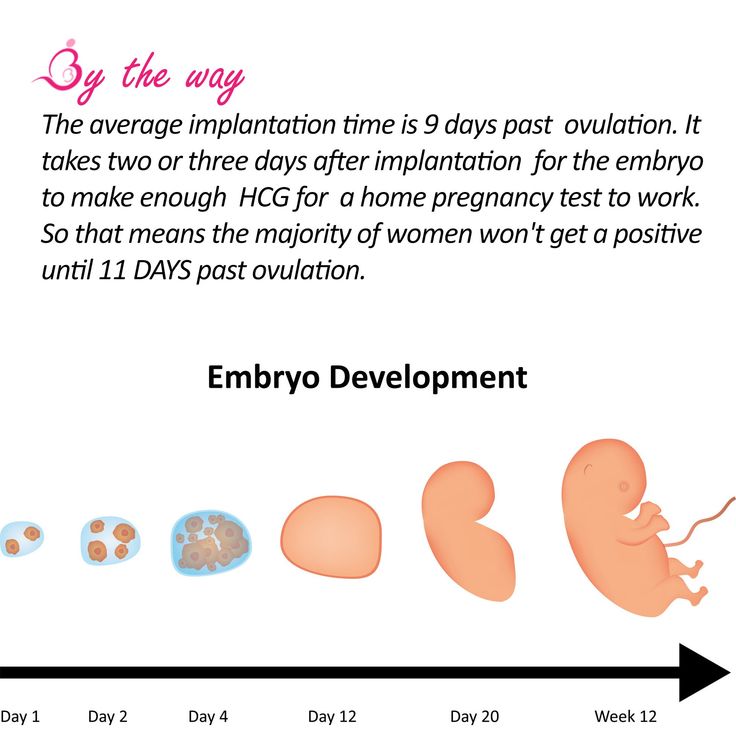 Still, existing prospective studies evaluating patient understanding and attitudes in such a screening program are limited in number, sample size, and diversity. A small, Internal Review Board-approved, pilot study was conducted at our institution with 100 women to ascertain their understanding of fragile X after genetic counseling and the motivations behind their prenatal screening choices. Seventy-two percent of the participants had never heard of fragile X prior to being counseled by the researcher. On a postcounseling quiz about fragile X syndrome and its inheritance pattern, 90% of the participants scored better than chance with a median score of 5 out of 6 true/false format questions answered correctly. Approximately 88% rated the counseling session as either helpful or extremely helpful. A total of 94% decided to undergo prenatal carrier screening for fragile X, with 54% selecting “I want to know” as their primary motivation.35
Still, existing prospective studies evaluating patient understanding and attitudes in such a screening program are limited in number, sample size, and diversity. A small, Internal Review Board-approved, pilot study was conducted at our institution with 100 women to ascertain their understanding of fragile X after genetic counseling and the motivations behind their prenatal screening choices. Seventy-two percent of the participants had never heard of fragile X prior to being counseled by the researcher. On a postcounseling quiz about fragile X syndrome and its inheritance pattern, 90% of the participants scored better than chance with a median score of 5 out of 6 true/false format questions answered correctly. Approximately 88% rated the counseling session as either helpful or extremely helpful. A total of 94% decided to undergo prenatal carrier screening for fragile X, with 54% selecting “I want to know” as their primary motivation.35
It is hoped that while reading the three clinical vignettes at the beginning of this article, prenatal screening for fragile X came to mind. In Case 1, DM’s nephew with mental retardation should prompt the clinician to investigate further and to ask whether a cause of the mental retardation was found. Unexplained mental retardation or developmental delay in a patient’s family history is an indication for prenatal testing. In addition, the clinician should inquire further about the demented grandfather who may have had signs of FXTAS. In Case 2, JP has a sister with early onset menopause that may be suggestive of FXPOI. JP should be offered screening because carrier status may alter a woman’s family-planning goals. JP may wish to preserve her fertility through methods such as oocyte cryopreservation or opt to have children earlier in life. Case 3 is more controversial as there are no clear indications for testing; however, failure to screen the donor eggs for fragile X could have grave consequences. RG did not inquire specifically about fragile X testing, but would like to know as much as possible about her potential pregnancy.
In Case 1, DM’s nephew with mental retardation should prompt the clinician to investigate further and to ask whether a cause of the mental retardation was found. Unexplained mental retardation or developmental delay in a patient’s family history is an indication for prenatal testing. In addition, the clinician should inquire further about the demented grandfather who may have had signs of FXTAS. In Case 2, JP has a sister with early onset menopause that may be suggestive of FXPOI. JP should be offered screening because carrier status may alter a woman’s family-planning goals. JP may wish to preserve her fertility through methods such as oocyte cryopreservation or opt to have children earlier in life. Case 3 is more controversial as there are no clear indications for testing; however, failure to screen the donor eggs for fragile X could have grave consequences. RG did not inquire specifically about fragile X testing, but would like to know as much as possible about her potential pregnancy.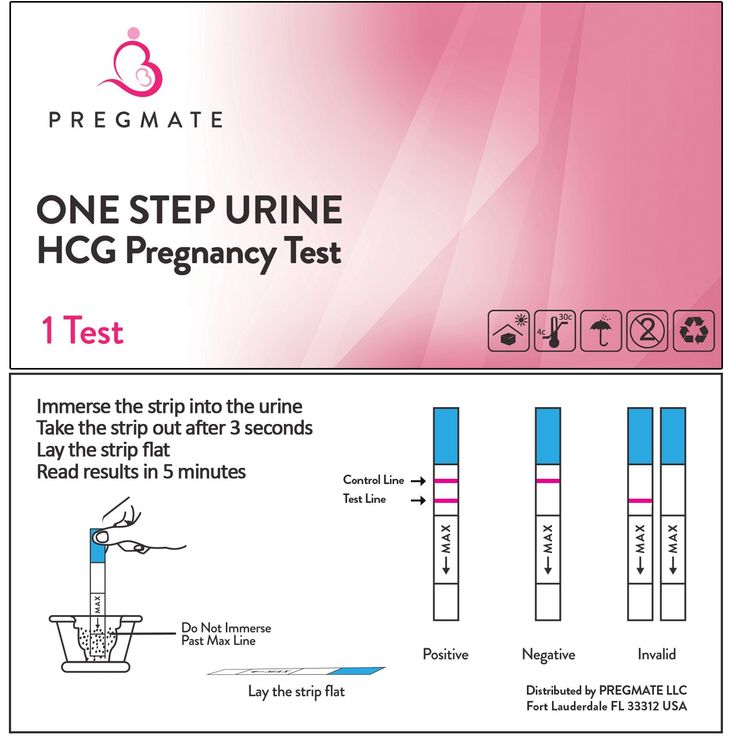 Current ACOG guidelines do not recommend screening in such cases, but many providers would still consider prenatal screening in this situation.
Current ACOG guidelines do not recommend screening in such cases, but many providers would still consider prenatal screening in this situation.
FMR1 gene mutations commonly result in inherited intellectual disability, infertility, and neurodegeneration syndromes that are encountered by clinicians in a variety of settings. Patients and clinicians are still largely unfamiliar with this disorder, its complicated inheritance, and its heterogeneous phenotype. Debate continues over who should be offered prenatal carrier screening. As more disease screening is offered, pretest counseling will only become more complex and clinicians will further struggle to balance the needs of the individual and allocation of public health resources. If universal prenatal screening is to be adopted, more research is needed regarding patient understanding of the tests they are undergoing, the psychological impact of such screening, and how this knowledge impacts health care and quality-of-life outcomes.
Main Points
Fragile X syndrome is the most common inherited form of intellectual and developmental disability worldwide. Unlike other causes of mental retardation such as Down syndrome, phenotypic features of fragile X often are not apparent until later in childhood, making it difficult to diagnose during the newborn period.
FMR1 gene mutations commonly result in inherited intellectual disability, infertility, and neurodegeneration syndromes that are encountered by clinicians in a variety of settings. Patients and clinicians are still largely unfamiliar with this disorder, its complicated inheritance, and its heterogeneous phenotype.
Debate continues over the individuals who should be offered prenatal carrier screening. Current guidelines from the American Congress of Obstetricians and Gynecologists recommend prenatal screening and genetic counseling for fragile X in women with a family or personal history of fragile X, unexplained mental retardation or developmental delay, or premature ovarian insufficiency.
As more disease screening is offered, pretest counseling will only become more complex and clinicians will continue to struggle to balance the needs of the individual and allocation of public health resources. If universal prenatal screening is to be adopted, more research is needed regarding patient understanding of the tests they are undergoing, the psychological impact of such screening, and how this knowledge impacts health care and quality-of-life outcomes.
1. Bailey DB , Jr, Raspa M, Bishop E, Holiday D. No change in the age of diagnosis for fragile X syndrome: findings from a national parent survey. Pediatrics. 2009;124:527–533. [PubMed] [Google Scholar]
2. American College of Medical Genetics, authors. Technical standards and guidelines for fragile X testing: a revision to the disease specific supplements to the Standards and guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics. Bethesda (MD): ACMG; 2005.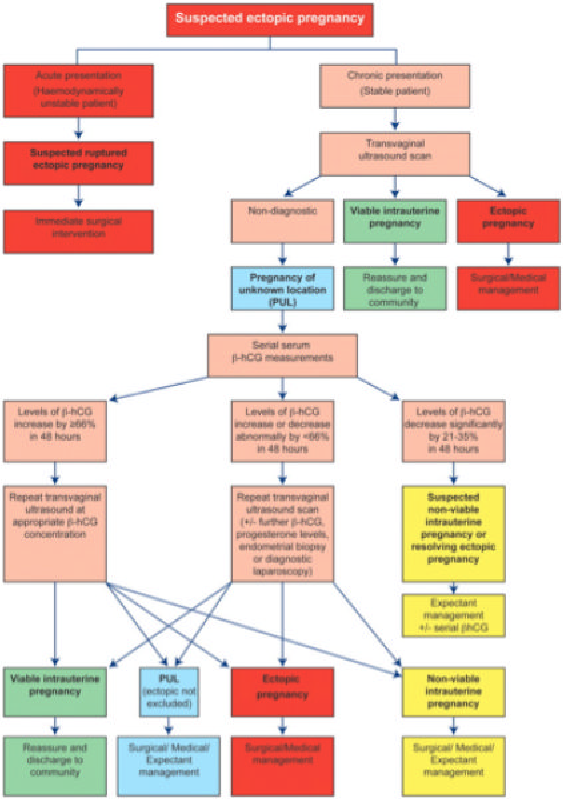 [Accessed August 2, 2012]. [Google Scholar]
[Accessed August 2, 2012]. [Google Scholar]
3. Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10:1038–1044. [PubMed] [Google Scholar]
4. Cronister A, Teicher J, Rohlfs EM, et al. Prevalence and instability of fragile X alleles: implications for offering fragile X prenatal diagnosis. Obstet Gynecol. 2008;111:596–601. [PubMed] [Google Scholar]
5. Tassone F, Hagerman RJ, Garcia-Arocena D, et al. Intranuclear inclusion in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J Med Genet. 2004;41:e43. [PMC free article] [PubMed] [Google Scholar]
6. Nolin SL, Brown WT, Glicksman A, et al. Expansion of the Fragile X CGG repeat in females with premutation or intermediate alleles. Am J Hum Genet. 2003;72:454–464. [PMC free article] [PubMed] [Google Scholar]
7. Gillberg C. Chromosomal disorders and autism. J Autism Dev Disord. 1998;28:415–425. [PubMed] [Google Scholar]
8. Hagerman RJ, Jackson AW 3rd, Levitas A, et al. An analysis of autism in fifty males with fragile X syndrome. Am J Med Genet. 1986;23:359–374. [PubMed] [Google Scholar]
Hagerman RJ, Jackson AW 3rd, Levitas A, et al. An analysis of autism in fifty males with fragile X syndrome. Am J Med Genet. 1986;23:359–374. [PubMed] [Google Scholar]
9. Hatton DD, Sideris J, Skinner M, et al. Autistic behavior in children with fragile X syndrome: prevalence, stability, and the impact of FMRP. Am J Med Genet A. 2006;140A:1804–1813. [PubMed] [Google Scholar]
10. Sherman S, Pletcher BA, Driscoll DA. Fragile X syndrome: diagnostic and carrier testing. Genet Med. 2005;7:584–587. [PMC free article] [PubMed] [Google Scholar]
11. Rogers SJ, Wehner EA, Hagerman R. The behavioral phenotype in fragile X: symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. Dev Behav Ped. 2001;22:409–417. [PubMed] [Google Scholar]
12. Schwartz CE, Dean J, Howard-Peebles PN, et al. Obstetrical and gynecological complications in fragile X carriers: a multicenter study. Am J Med Genet. 1994;51:400–402. [PubMed] [Google Scholar]
13. Sherman SL. Premature ovarian failure in the fragile X syndrome. Am J Med Genet. 2000;97:189–194. [PubMed] [Google Scholar]
Sherman SL. Premature ovarian failure in the fragile X syndrome. Am J Med Genet. 2000;97:189–194. [PubMed] [Google Scholar]
14. Karimov CB, Moragianni VA, Cronister A, et al. Increased frequency of occult fragile X-associated primary ovarian insufficiency in infertile women with evidence of impaired ovarian function. Hum Reprod. 2011;26:2077–2083. [PubMed] [Google Scholar]
15. Jacquemont S, Hagerman RJ, Leehey MA, et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in premutation carrier population. JAMA. 2004;291:460–469. [PubMed] [Google Scholar]
16. Leehey MA. Fragile X-associated tremor/ataxia syndrome: clinical phenotype, diagnosis and treatment. J Invest Med. 2009;57:830–836. [PMC free article] [PubMed] [Google Scholar]
17. Brunberg JA, Jacquemont S, Hagerman RJ, et al. Fragile X premutation carriers: characteristic MR imaging findings of adult male patients with progressive cerebellar and cognitive dysfunction. Am J Neuroradiol. 2002;23:1757–1766. [PMC free article] [PubMed] [Google Scholar]
[PMC free article] [PubMed] [Google Scholar]
18. American College of Obstetricians and Gynecologists Committee on Genetics, authors. ACOG Committee Opinion No. 469: Carrier screening for fragile X syndrome. Obstet Gynecol. 2010;116:1008–1010. [PubMed] [Google Scholar]
19. Hill MK, Archibald AD, Cohen J, Metcalfe SA. A systematic review of population screening for fragile X syndrome. Genet Med. 2010;12:396–410. [PubMed] [Google Scholar]
20. Finucane B. Should all pregnant women be offered carrier testing for fragile X syndrome? Clin Obstet Gynecol. 1996;39:772–782. [PubMed] [Google Scholar]
21. Rajendra K, Bringman JJ, Ward J, Phillips OP. Who should be tested for fragile X carriership? A review of one center’s pedigrees. Am J Obstet Gynecol. 2008;198:e51–e53. [PubMed] [Google Scholar]
22. Acharya K, Ross LF. Fragile X screening: attitudes of genetic health professionals. Am J Med Genet. 2009;149A:626–632. [PMC free article] [PubMed] [Google Scholar]
23. McConkie-Rosell A, Spiridigliozzi GA, Iafolla T, et al. Carrier testing in the fragile X syndrome: attitudes and opinions of obligate carriers. Am J Med Genet. 1997;68:62–69. [PubMed] [Google Scholar]
McConkie-Rosell A, Spiridigliozzi GA, Iafolla T, et al. Carrier testing in the fragile X syndrome: attitudes and opinions of obligate carriers. Am J Med Genet. 1997;68:62–69. [PubMed] [Google Scholar]
24. Skinner D, Sparkman KL, Bailey DB., Jr Screening for fragile X syndrome: parent attitudes and perspectives. Genet Med. 2003;5:378–384. [PubMed] [Google Scholar]
25. Musci TJ, Caughey AB. Cost-effectiveness analysis of prenatal population-based fragile X carrier screening. Am J Obstet Gynecol. 2005;192:1905–1915. [PubMed] [Google Scholar]
26. Wildhagen MF, van Os TA, Polder JJ, et al. Explorative study of costs, effects and savings of screening for female fragile X premutation and full mutation carriers in the general population. Community Genet. 1999;1:36–47. [PubMed] [Google Scholar]
27. Ram KT, Klugman SD. Best practices: antenatal screening for common genetic conditions other than aneuploidy. Curr Opin Obstet Gynecol. 2010;22:139–145. [PubMed] [Google Scholar]
28. Sayres LC, Cho MK. Cell-free fetal nucleic acid testing: a review of the technology and its applications. Obstet Gynecol Surv. 2011;66:431–442. [PubMed] [Google Scholar]
Sayres LC, Cho MK. Cell-free fetal nucleic acid testing: a review of the technology and its applications. Obstet Gynecol Surv. 2011;66:431–442. [PubMed] [Google Scholar]
29. Tsui HN, Lo YM. Recent advances in the analysis of fetal nucleic acids in maternal plasma. Curr Opin Hematol. 2012;19:462–468. [PubMed] [Google Scholar]
30. Allyse M, Sayres LC, King JS, et al. Cell-free fetal DNA testing for fetal aneuploidy and beyond: clinical integration challenges in the US context. Hum Reprod. 2012;27:3123–3131. [PMC free article] [PubMed] [Google Scholar]
31. Finucane B, Abrams L, Cronister A, et al. Genetic counseling and testing for the FMR1 gene mutations: practice guidelines of the National Society of Genetic Counselors. J Genet Counsel. 2012;21:752–760. [PubMed] [Google Scholar]
32. Nolin SL, Sah S, Glicksman A, et al. Fragile X AGG analysis provides new risk predictions for 45-69 repeat alleles. Am J Med Genet. doi:10.1002/ajmg.a.3583. [PMC free article] [PubMed] [Google Scholar]
33. Nolin SL, Glicksman A, Ding X, et al. Fragile X analysis of 1112 prenatal samples from 1991 to 2010. Prenat Diagn. 2011;31:925–931. [PubMed] [Google Scholar]
Nolin SL, Glicksman A, Ding X, et al. Fragile X analysis of 1112 prenatal samples from 1991 to 2010. Prenat Diagn. 2011;31:925–931. [PubMed] [Google Scholar]
34. Musci TJ, Moyer KM. Prenatal carrier testing for fragile X: counseling issues and challenges. Obstet Gynecol Clin North Am. 2012;37:61–70. [PubMed] [Google Scholar]
35. Gutiérrez JF, Klugman SD, Bajaj K. Fragile X carrier screening: knowledge, practice and attitudes in a diverse urban population; Presented at the American College of Medical Genetics Annual Meeting; March 20; Vancouver, BC. [Google Scholar]
Fragile X-chromosome analysis (FMR - repeat) in Nizhny Novgorod
I applied for a preventive examination, I liked the communication with Dr. Koroleva K.V. A very attentive doctor, she explained everything, calmed down. They measured the eye pressure, gave recommendations. Thank you. There was no queue, the appointment was made by appointment.
Olga
I have been seeing a gynecologist Viktoria Orlova for about 3 years now. It's a miracle! I moved to another city, but still every time I guess, so that upon arrival in Nizhny I come for a scheduled inspection. And how many times I needed to consult, I probably won’t count - the doctor is always on the phone and there were force majeure when I don’t know what I would do without Viktoria Vladimirovna. And I just have to say to those who are afraid to do the HSG (just like I was afraid after reading articles on the Internet) - it does not hurt at all. The stomach will pull for a few minutes, as in the first days of menstruation, and that's it. Taking a normal smear is even more unpleasant) I can’t speak for other doctors of the clinic - I am faithful to my doctor and have never been to anyone else), but I vouch for my doctor. In recent years, I feel calm and confident in terms of women's health thanks to V.V. Orlova. And this is the only doctor out of five in other clinics in the city that I went to, who was able to pick up such hormones for me, the consequences of which I don’t feel - I don’t go crazy, I don’t get fat.
It's a miracle! I moved to another city, but still every time I guess, so that upon arrival in Nizhny I come for a scheduled inspection. And how many times I needed to consult, I probably won’t count - the doctor is always on the phone and there were force majeure when I don’t know what I would do without Viktoria Vladimirovna. And I just have to say to those who are afraid to do the HSG (just like I was afraid after reading articles on the Internet) - it does not hurt at all. The stomach will pull for a few minutes, as in the first days of menstruation, and that's it. Taking a normal smear is even more unpleasant) I can’t speak for other doctors of the clinic - I am faithful to my doctor and have never been to anyone else), but I vouch for my doctor. In recent years, I feel calm and confident in terms of women's health thanks to V.V. Orlova. And this is the only doctor out of five in other clinics in the city that I went to, who was able to pick up such hormones for me, the consequences of which I don’t feel - I don’t go crazy, I don’t get fat. And until I came to the clinic, I took a lot of pills
And until I came to the clinic, I took a lot of pills
Svetlana
On June 16, I had a consultation with a cardiologist Larisa Mikhailovna Lokonova. I left with the full feeling that this is how all doctors should treat you: they will listen to you, carefully look at all your tests, give competent advice, recommend treatment. Thanks to Larisa Mikhailovna, people like her strengthen faith in our medicine.
Valentina June 23, 2022
I thank your center and doctor Yulia Valerievna Gogleva for prompt help! Recently, I had an eye injury, and during the examination after it, they found that the retina is very thin, I was lucky that the injury did not lead to its rupture. Nevertheless, I was strongly recommended to strengthen it with a laser, because after some time, especially given the injury, it could begin to tear. Yulia Valerievna showed me all the pictures, clearly explained how things were going, and performed a laser operation on me. By the way, it is quite comfortable to carry. Thanks again for your timely help! nine0003
Thanks again for your timely help! nine0003
Sergey
Hello! In April, I went through examinations, as I am pregnant, and also went to the ophthalmologist, and I was diagnosed with retinal dystrophy. Accordingly, there was a great danger of her rupture during childbirth, this issue had to be resolved. I turned to your doctor Terentyeva A.A., and at the appointment she told me that special laser surgeries are being carried out to strengthen the retina (which I didn’t even know about before), and that in my case the operation would remove the risk of such a rupture. Of course, I did not think long about making a decision, and I came to A.A. on this laser reinforcement. I did an operation on my eyes for the first time, and I didn’t even think that I would encounter this, I was very worried. But I was surprised how simple the operation was, I imagined everything much more painful and longer :) As a result, after the operation, my retina was checked again, and there is no longer a risk of damaging it during childbirth. Thank you very much to your doctor and your clinic for helping me finally resolve this issue, because of which I was really very nervous)
Thank you very much to your doctor and your clinic for helping me finally resolve this issue, because of which I was really very nervous)
Olga
In your clinic, we performed laser cataract surgery on my father. Thanks to your doctor - Dozorova Irina Pavlovna for the approach and quality of the operation. Everything was done quickly and clearly. The father no longer complains about his vision, he sees well. Your center was recommended by a friend of my wife, and we were very pleased with the choice.
Mikhail
A little over a month ago we faced a problem - my grandmother lost her eyesight after cataract surgery, which we performed on her 4 years ago. She was diagnosed with a secondary cataract and needed surgery. Your clinic was advised to us by an ophthalmologist at the clinic, and we decided not to look for other options, especially since the operation had to be carried out as quickly as possible, my grandmother began to see really very badly. We got an appointment with E. N. Shabalina, a very attentive doctor, I also noted a good attitude towards my grandmother, because she herself was very nervous and afraid of doctors, but everything went well. At the reception, Elena Nikolaevna appointed the day of the operation in about a week. The operation was laser, so it was easy, now my vision has fully recovered, there are no complaints from my grandmother, and we are glad that we nevertheless turned to you! nine0003
N. Shabalina, a very attentive doctor, I also noted a good attitude towards my grandmother, because she herself was very nervous and afraid of doctors, but everything went well. At the reception, Elena Nikolaevna appointed the day of the operation in about a week. The operation was laser, so it was easy, now my vision has fully recovered, there are no complaints from my grandmother, and we are glad that we nevertheless turned to you! nine0003
Nika
Thanks to your clinic and your doctor Irina Pavlovna for helping my mother regain normal vision!!! It worsened so much that we could not even pick up glasses, they simply stopped helping, it turned out that everything was due to cataracts. Irina Pavlovna performed the operation, the doctor herself is very friendly, it was very pleasant at her appointments. Mom is overjoyed that she can see well again, and that it was not so difficult to achieve this!
Inga
2 weeks ago in your clinic my father was operated on by doctor Shabalina Elena Nikolaevna. We are very pleased with the work of the doctor, the operation was successful, the father noted that during the operation he practically felt nothing, there was no pain, and the result was excellent. At the reception before the operation, Elena Nikolaevna spoke in detail about how the procedure would go, what kind of lens it would be (we decided to do it for a fee, but I know that there is also an opportunity to do it for free at the clinic), so we were prepared. Vision has improved significantly, the father stopped complaining about "blurring" and dull colors, he began to see as before, thank you very much! nine0003
We are very pleased with the work of the doctor, the operation was successful, the father noted that during the operation he practically felt nothing, there was no pain, and the result was excellent. At the reception before the operation, Elena Nikolaevna spoke in detail about how the procedure would go, what kind of lens it would be (we decided to do it for a fee, but I know that there is also an opportunity to do it for free at the clinic), so we were prepared. Vision has improved significantly, the father stopped complaining about "blurring" and dull colors, he began to see as before, thank you very much! nine0003
Roman
Finally, we were able to solve the issue of my grandmother's cataract operation in a timely manner and even for free (!)! They performed the operation under the OMS policy, and it was unexpectedly pleasant that, starting from a conversation with the administrator and ending with the operation itself, everything was carried out so efficiently, quickly and politely. Initially, grandma's eyesight began to deteriorate very much, and it was necessary to urgently resolve this issue, we did not even immediately know what was the matter, your ophthalmologist Lyudmila Gennadievna consulted us and signed up for an operation, which she performed, we waited in line not very long. I am very glad that the first time we were able to find a medical center where we were provided with all the necessary assistance and we did not waste time! nine0003
Initially, grandma's eyesight began to deteriorate very much, and it was necessary to urgently resolve this issue, we did not even immediately know what was the matter, your ophthalmologist Lyudmila Gennadievna consulted us and signed up for an operation, which she performed, we waited in line not very long. I am very glad that the first time we were able to find a medical center where we were provided with all the necessary assistance and we did not waste time! nine0003
Julia
Fragile X syndrome
Fragile X syndrome
(FXS, Martin-Bell syndrome)
Fragile X Syndrome (FXS, Martin-Bell Syndrome)
This is an inherited X-linked (sex-linked) disorder resulting from a disorder in the FMR1 gene (fragile X mental retardation 1). The syndrome is characterized by mental retardation of varying degrees, impaired concentration, speech development, and may also be accompanied by other clinical manifestations. nine0003
Fragile X Syndrome can occur in both sexes, but men suffer from this disease more often and more severely, as they have only one X chromosome.
Prevalence: 1 in 4000 men and 1 in 6000 women.
What does "fragility" of the
X-chromosome mean?
The "fragility" of the X chromosome lies in its special structure.
In the course of cytogenetic studies, it was found that when folic acid was introduced into the cell culture of some patients with mental retardation, a small percentage of cells on the long arm of one of the X chromosomes, in which the mutated gene FMR1 , the edge seems to be laced up, hanging on a thread.
However, this method is not sensitive enough and is currently rarely used to diagnose the syndrome.
The genetic basis of this phenomenon is an increase in the number of trinucleotide repeats (that is, an increase in the number of CGG nucleotides) on the long arm of the X chromosome.
In healthy people, the number of these repeats ranges from 6 to 45. The clinical manifestation of the disease is observed when the number of CGG repeats is over 200, which is considered a “mutation”. The state when the number of CGG repeats is in the range of 55-200 is called "premutation". People with this many CGG repeats do not have the disease in its typical form, but it is highly likely that it will manifest itself in their descendants. This is due to the possibility of an increase in the number of trinucleotide repeats during oogenesis. nine0003
The state when the number of CGG repeats is in the range of 55-200 is called "premutation". People with this many CGG repeats do not have the disease in its typical form, but it is highly likely that it will manifest itself in their descendants. This is due to the possibility of an increase in the number of trinucleotide repeats during oogenesis. nine0003
"Gray zone" is the number of repeats from 45 to 54. It has no effect on human health, but can lead to problems in future generations, as in the case of a premutational state of the gene.
Clinical features of Fragile X Syndrome
Patients with this syndrome have intellectual disabilities ranging from moderate learning disabilities to more severe problems. They are also characterized by fast, confused and muttering speech. nine0003
Boys have characteristic phenotypic (facial) features: a large head with a high and wide forehead, a long face with an enlarged chin, a somewhat flattened middle part of the face, a blunt, slightly beak-like curved tip of the nose and large protruding ears.
Behavioral characteristics of may include autism and autistic traits, hyperactivity, hand-biting and clapping, aggressiveness, difficult eye contact. The pathognomonic sign in postpubertal age, which makes one suspect the disease, is macroorchism (increased testicle size) in the absence of endocrine pathology.
Other characteristic manifestations are joint hypermobility, frequent otitis, strabismus, mitral valve prolapse, gastroesopharyngeal reflux. The clinical picture can vary greatly, there may be only one or several signs.
The main symptoms observed in men can also be observed in women. However, they are dominated by milder intellectual disabilities, mild behavioral problems, and physical signs of FXS. nine0058
Approximately one third of women with FXS are severely mentally retarded, while the rest may have moderate or some learning disabilities, emotional, mental problems, general anxiety and/or social anxiety.
A small percentage of women with a complete mutation in the FMR1 gene do not have obvious signs of intellectual, behavioral or physical abnormalities. FXS is often diagnosed in them only after the disease is found in other family members. nine0003
FXS is often diagnosed in them only after the disease is found in other family members. nine0003
Disorders associated with
premutation (55-200 CGG repeats) in the FMR1 gene
Primary ovarian failure (FXPOI), associated with fragile X chromosome
Primary ovarian failure is characterized by decreased ovarian reserve, menopause before age 40, and increased levels of follicle-stimulating hormone (FSH).
The term "primary ovarian insufficiency" or "premature ovarian failure" (POF) is currently used to define a disease that was previously characterized as premature menopause, premature ovarian failure, or secondary hypogonadal gonadism. One of the main hallmarks of premature ovarian failure is an increase in the level of follicle-stimulating hormone. Most cases of POI are sporadic, with an average incidence of about 9 in women with a chromosome set of 46 XX.0057 1% , while a close relationship of this disorder with age has been proven.
However, approximately 10-15% of patients with POI have a positive family history.
Two main mechanisms can be identified in the etiology of POI: reduction in the number (pool depletion) of follicles and their dysfunction. The reasons leading to the development of POI are very heterogeneous: genetic, enzymatic, autoimmune, infectious-toxic, iatrogenic, psychogenic. In recent years, much attention has been paid by researchers to the molecular genetic aspects of this ovarian pathology, since a certain set of genes has been identified that may be responsible for the development of POF. It has been shown that changes in gene FMR1 , located on the X chromosome (Xq26-28 region), significantly increase the risk of this pathology.
Fragile X primary ovarian failure (FXPOI) occurs in carriers of the premutation (55-200 CGG repeats). In women with sporadic forms of POF, the frequency of premutation of the FMR1 gene ranges from 0.8 to 7. 5% , while in familial forms of the disease it can reach 13% . It has been shown that female carriers of gene 9 premutation0051 FMR1 are much more likely (38%) to suffer from oligomenorrhea compared to the general population data (6%).
5% , while in familial forms of the disease it can reach 13% . It has been shown that female carriers of gene 9 premutation0051 FMR1 are much more likely (38%) to suffer from oligomenorrhea compared to the general population data (6%).
In this case, the presence of premutational alleles (~60-200 repeats) in a woman, there is a risk of an increase in the number of repeats (expansion) in her descendants to a full mutation, which increases with the size of the repeat region. When the number of repeats exceeds 100, there is a systematic risk of expansion to full mutation in the next generation.
Typically maternal premutation of gene FMR1 goes into a full mutation in the offspring, resulting in fragile X syndrome, while the paternal premutation is usually stable. Many FMR1 -alleles contain AGG sequences scattered among the CGG repeat regions. These AGGs are thought to form "breaks" that confer stability on the DNA and reduce the risk of expansion into the next generation. The risk profile of mothers without AGG breaks is higher than that of mothers with the same number of repeats but with at least one AGG region that reduces the number of contiguous (CGG)n sequences. nine0003
The risk profile of mothers without AGG breaks is higher than that of mothers with the same number of repeats but with at least one AGG region that reduces the number of contiguous (CGG)n sequences. nine0003
Tremor/ataxia, associated with fragile X chromosome (FXTAS)
Premutation also correlates with manifestation of fragile X-associated tremor-ataxic syndrome (FXTAS).
FXTAS is an older neurodegenerative disease that is more common in men. Its first symptoms usually appear after 50 years of age. Prior to the onset of symptoms of FXTAS, most patients did not have any medical or neurological problems. Women make up only a small proportion of FXTAS patients, and their symptoms tend to be less severe. nine0003
Features of FXTAS include:
- Cerebellar ataxia (balance problems).
- Intentional tremor (dynamic movements that appear or increase during some actions, writing, etc.).
- Memory loss (usually short term).
- Mood instability, irritability, personality change, psychiatric symptoms.
- Symptoms of parkinsonism (many patients are misdiagnosed with Parkinson's disease). nine0178
- Dementia (misdiagnosed as Alzheimer's).
- Cognitive decline (decrease in math, reading and verbal skills, comprehension).
- FXTAS progression at different rates.
Genetics and inheritance
As mentioned earlier, in 99% of cases, the root cause of FXS is an increase in the number of CGG tandem triplet repeats in the 5'-untranslated region of the FMR1 gene . (Other mutations in and around the same gene and mutations in the close homologue FMR2 have been reported to produce a similar phenotype, but this is very rare.) In all humans, the FMR1 gene contains CGG tandem triplet repeats in the 5' untranslated region. When a gene is passed on to offspring, this number may slightly decrease or increase: in generations, this section of the genome, as it were, “breathes”.
If a woman has more than 50 repeats in one of her X chromosomes (statistically, this occurs in one in 250 X chromosomes, that is, very often), there is a risk of a sharp spontaneous increase in oocyte progenitor cells during meiosis. nine0058
If, as a result of such a spontaneous increase, the number of repeats exceeds 200 and it is this allele that the child gets, this allele FMR1 will not be expressed at all in him.
If this child turns out to be a girl, then the effect of the deletion can be compensated to some extent by the work of the normal allele on the X chromosome received from the father, especially with a favorable pattern of inactivation of one of the X chromosomes in somatic cells.
If the child is a boy, then he will develop FXS in all its severity. nine0003
The expansion of the number of repeats manifests itself depending on its degree
Carrying the so-called premutation (number of repeats from 50 to 200) may not be expressed phenotypically in any way, since the FMR1 gene is still expressed, or, as was described above leads to FXPOI in women of reproductive age and to FXTAS in men and women over 50 years of age.
With a complete mutation (the number of repeats is more than 200, the gene is not expressed: the cellular systems react to the extensive expansion of CGG repeats by methylating the promoter, i.e. the upstream DNA region where gene transcription is initiated. Due to the blocking of the gene 9 promoter0051 FMR1 not expressed, FMRP protein not synthesized; it does not fulfill its function.
Normally, the FMRP protein is present in various organs and tissues, but is especially pronounced in the nervous system. The FMRP protein is the most important regulator of mRNA processing in nerve cells. Disabling expression of FMR1 due to a critical increase in the number of CGG repeats in the 5'-untranslated region of the gene changes the entire profile of protein synthesis in brain neurons. At the level of cellular morphology, this is expressed in the formation of an abnormally large number of dendritic spines of an altered shape. nine0003
Arising in large numbers, such spines do not produce functionally mature synapses; the result is an anomalous hyperplasticity of neural networks, incompatible with the reception and processing of information from the outside world in a normal mode characteristic of a person.
Fragile X syndrome can be diagnosed by the behavior of the child by the end of the first year of life, but in the first weeks and even months, this is usually impossible, i.e. the syndrome is not "obstetric".
Pedigrees of patients with fragile X syndrome clearly show the phenomenon of anticipation. This phenomenon consists in the aggravation of clinical manifestations in each new generation due to the increase in the expansion of trinucleotide repeats.
In this regard, screening and diagnosis of fragile X syndrome are of great clinical importance.
Who should be diagnosed for
fragile X syndrome?
Genetic test for FMR1 gene disorders ,
required:
- Patients with developmental delay, intellectual disability of unknown etiology or autism and their relatives.
- Women with reproductive disorders associated with elevated FSH levels, oligomenorrhea.
- Patients with tremor and cerebellar ataxia in the elderly.

- Patients whose relatives have been found to have abnormalities in the FMR1 gene.
nine0048 How is Fragile X syndrome tested
The main method in the diagnosis of FXS, FX-TAS and FX-POI, as well as to identify the risk of expansion of repeats to full mutation in the next generation in patients with premutations, is the polymerase chain reaction (PCR) with the detection of the obtained products by capillary electrophoresis.
The assay provides premutation detection by accurate allele sizing of up to 200 CGG repeats and identification of full mutational CGG alleles in both males and females, and allows identification of AGG breaks. The conditions for this test are listed here. nine0003
Basic principles
Diagnostics
Genospecific PCR
CGG RP PCR
Data Electrophoregrams of Panel Panel (NIBSMO (NIBS
Electrophoregram demonstrating the presence of AGG breaks in alleys 30 and 60 CGG
Why diagnose this
syndrome if it is not treated?
From the point of view of reproductive medicine, it is very important to be able to predict the birth of new children with FXS.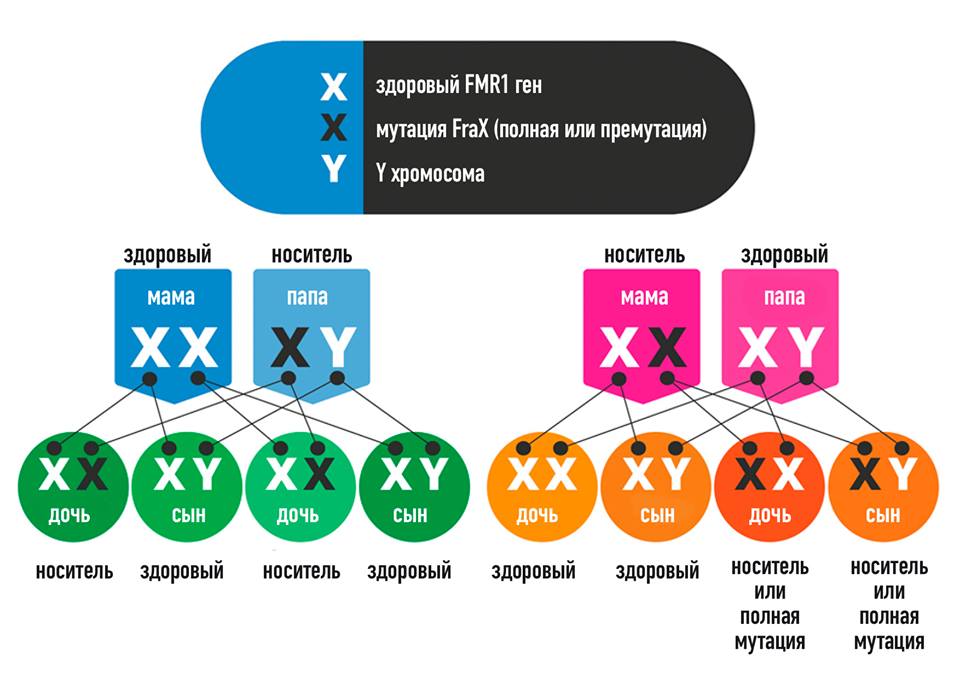 Now it's possible!
Now it's possible!
From the point of view of corrective practice, work with such children can be improved: recently, clinical trials have been completed in the United States of drugs that can alleviate a number of symptoms, in particular, improve socialization (Hagermanetal, 2012).
Why pregnant women should be screened (as early as possible) rather than neonates (in developed countries testing for the risk of FXS in offspring is already included in many pregnancy management programs)?
Firstly, if a premutation is detected in a potential woman in labor, it is still possible to make sure that the “sick” chromosome does not get to the child at all (a woman has two X chromosomes, she can pass on the “healthy” one to the child).
For this, there is IVF (or perinatal diagnosis in the early stages, if the scenario is unsuccessful, you can terminate the pregnancy). nine0058
Women tested (n= 14,334)
Normal allele size
((n= 14,127)
Premutation
(50 -199)
(n= 204)
Full mutation
(>200)
(n= 3)
Pregnancies (n= 193)
Amniocentesis/chorionic villus sampling
(n= 177)
Normal
fetal allele
(n= 87)
Premutation
fetal allele
(n= 85)
Fully mutated
fetal allele
(n= 5)
H. Toledano-Alhadefetal., AmJHumGenet 69:351–360, 2001
Toledano-Alhadefetal., AmJHumGenet 69:351–360, 2001
Second, from the point of view of molecular biology methods with their limitations, detection of a premutation is easier and more reliable than a full mutation. Premutation is better detected by PCR, while a full mutation >500 repeats may not give a PCR product (a girl with a full mutation on one of the X chromosomes may look like she has the same normal number of repeats on both chromosomes ; compare the third and sixth samples in the electrophoresis diagram):
Number of CGG repeats
What to do after receiving
results?
According to the results of DNA diagnostics, it is necessary to consult a geneticist, who will be able to correctly interpret the results and draw up a plan for further management of the patient.
If an asymptomatic carrier of disorders in the FMR1 gene is detected, family planning issues and current opportunities for having a healthy child will be discussed.